Freeze-Thaw-Induced Embolism and Ultrasonic Emissions in Angiosperms
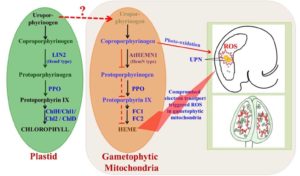 All organisms including plants share the tetrapyrrole biosynthesis pathway that is critical for the production of compounds such as heme and chlorophyll. During tetrapyrrole biosynthesis, coproporphyrinogen III oxidase (CPO) catalyzes the conversion of coproporphyrinogen III into protoporphyrinogen IX. Pratibha et al. () report the results of the characterization of a mutation in the Arabidopsis gene At5g63290 that is orthologous to bacterial and mammalian CPO. As this gene shows greater homology with HemN-like CPO, they named it AtHEMN1. Loss of AtHEMN1 function increased coproporphyrinogen III level and reduced protoporphyrinogen IX level, suggesting the impairment of tetrapyrrole biosynthesis. Mutations that disrupted AtHEMN1 adversely affected silique length, ovule number, and seed set. Athemn1 mutant alleles could be transmitted via both male and female gametes, but homozygous mutants were never recovered. Embryo development in Athemn1 was arrested at the globular stage but the mutant phenotype was completely rescued by transgenic expression of AtHEMN1. Promoter and transcript analyses indicated that AtHEMN1 is expressed mainly in floral tissues and developing seeds. AtHEMN1-green fluorescent protein fusion protein was found targeted to mitochondria. Athemn1 mutant alleles also showed defects in gametophyte development, including nonviable pollen and embryo sacs with unfused polar nuclei. Improper differentiation of the central cell led to defects in endosperm development. Inactivation of any of the enzymes of the tetrapyrrole biosynthetic pathway leads to the accumulation of porphyrin compounds and causes cell death in plants through reactive oxygen species (ROS) production. As expected, the blockage of tetrapyrrole biosynthesis in the Athemn1 mutant also led to increased ROS accumulation in anthers and embryo sacs, as evidenced by nitroblue tetrazolium staining. Thus, it appears that that the tetrapyrrole/heme biosynthesis pathway operates in mitochondria and its impairment disturbs ROS homeostasis in flower buds, thereby adversely affecting male and female gametophyte development in Arabidopsis.
All organisms including plants share the tetrapyrrole biosynthesis pathway that is critical for the production of compounds such as heme and chlorophyll. During tetrapyrrole biosynthesis, coproporphyrinogen III oxidase (CPO) catalyzes the conversion of coproporphyrinogen III into protoporphyrinogen IX. Pratibha et al. () report the results of the characterization of a mutation in the Arabidopsis gene At5g63290 that is orthologous to bacterial and mammalian CPO. As this gene shows greater homology with HemN-like CPO, they named it AtHEMN1. Loss of AtHEMN1 function increased coproporphyrinogen III level and reduced protoporphyrinogen IX level, suggesting the impairment of tetrapyrrole biosynthesis. Mutations that disrupted AtHEMN1 adversely affected silique length, ovule number, and seed set. Athemn1 mutant alleles could be transmitted via both male and female gametes, but homozygous mutants were never recovered. Embryo development in Athemn1 was arrested at the globular stage but the mutant phenotype was completely rescued by transgenic expression of AtHEMN1. Promoter and transcript analyses indicated that AtHEMN1 is expressed mainly in floral tissues and developing seeds. AtHEMN1-green fluorescent protein fusion protein was found targeted to mitochondria. Athemn1 mutant alleles also showed defects in gametophyte development, including nonviable pollen and embryo sacs with unfused polar nuclei. Improper differentiation of the central cell led to defects in endosperm development. Inactivation of any of the enzymes of the tetrapyrrole biosynthetic pathway leads to the accumulation of porphyrin compounds and causes cell death in plants through reactive oxygen species (ROS) production. As expected, the blockage of tetrapyrrole biosynthesis in the Athemn1 mutant also led to increased ROS accumulation in anthers and embryo sacs, as evidenced by nitroblue tetrazolium staining. Thus, it appears that that the tetrapyrrole/heme biosynthesis pathway operates in mitochondria and its impairment disturbs ROS homeostasis in flower buds, thereby adversely affecting male and female gametophyte development in Arabidopsis.
 Plant-parasitic cyst nematodes (Heterodera species) are among the most devastating pathogens of plant roots. These obligate parasites initiate a long period of biotic interactions with their host plants where formation of an operative feeding structure, the syncytium, is vital for nematode survival and development. The nematode provokes differentially terminated cells in the vascular root tissues to redifferentiate into a syncytium cell type, a switch that involves simultaneous changes in the expression of thousands of genes. Though the mechanisms controlling gene expression changes in the syncytium remain ill defined, recent studies indicate that epigenetic mechanisms including noncoding small RNAs and DNA methylation may play fundamental roles. DNA methylation can regulate the expression of protein-coding genes and the activity of transposable elements. Hewezi et al. (
Plant-parasitic cyst nematodes (Heterodera species) are among the most devastating pathogens of plant roots. These obligate parasites initiate a long period of biotic interactions with their host plants where formation of an operative feeding structure, the syncytium, is vital for nematode survival and development. The nematode provokes differentially terminated cells in the vascular root tissues to redifferentiate into a syncytium cell type, a switch that involves simultaneous changes in the expression of thousands of genes. Though the mechanisms controlling gene expression changes in the syncytium remain ill defined, recent studies indicate that epigenetic mechanisms including noncoding small RNAs and DNA methylation may play fundamental roles. DNA methylation can regulate the expression of protein-coding genes and the activity of transposable elements. Hewezi et al. (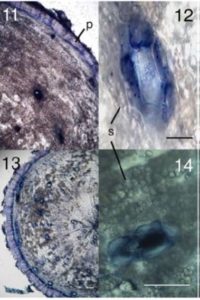 The Mediterranean plant Thapsia garganica (Apiaceae), also known as deadly carrot, produces the highly toxic compound thapsigargin. This compound is a potent inhibitor of the sarcoplasmic-endoplasmic reticulum Ca2+-ATPase calcium pump in mammals and is of industrial importance as the active moiety of the anticancer drug mipsagargin, currently in clinical trials. Thapsigargin is found in most parts of the plant T. garganica. Ripe fruits contain the highest amount of thapsigargin, with 0.7% to 1.5% of the dry weight, followed by roots (0.2%–1.2% of dry weight) and leaves (0.1% of dry weight). It is well established that many Apiaceae species store lipophilic compounds such as phenyl propanoids and terpenoids in secretory ducts, and this appears to be the case with T. gargantica as well. Andersen et al. (
The Mediterranean plant Thapsia garganica (Apiaceae), also known as deadly carrot, produces the highly toxic compound thapsigargin. This compound is a potent inhibitor of the sarcoplasmic-endoplasmic reticulum Ca2+-ATPase calcium pump in mammals and is of industrial importance as the active moiety of the anticancer drug mipsagargin, currently in clinical trials. Thapsigargin is found in most parts of the plant T. garganica. Ripe fruits contain the highest amount of thapsigargin, with 0.7% to 1.5% of the dry weight, followed by roots (0.2%–1.2% of dry weight) and leaves (0.1% of dry weight). It is well established that many Apiaceae species store lipophilic compounds such as phenyl propanoids and terpenoids in secretory ducts, and this appears to be the case with T. gargantica as well. Andersen et al. (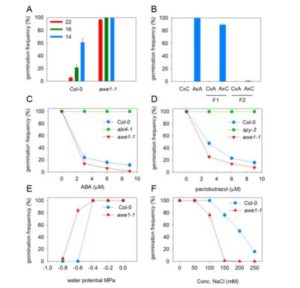 Environmental signals during seed production are important determinants of seed properties, including seed dormancy and seed longevity. The mother plant plays an important role in this signaling process, collecting signals throughout her life history and modulating dormancy by providing hormones to maturing seeds and by plastic development of the tissues surrounding the embryo. This process is especially important in seeds with physiological dormancy that is coat imposed, which requires the presence and activity of the seed coat and endosperm structures that form a barrier between the embryo and the external environment. In order to further understand the mechanisms by which the control of coat-induced dormancy takes place in Arabidopsis (Arabidopsis thaliana), Fedi et al. (
Environmental signals during seed production are important determinants of seed properties, including seed dormancy and seed longevity. The mother plant plays an important role in this signaling process, collecting signals throughout her life history and modulating dormancy by providing hormones to maturing seeds and by plastic development of the tissues surrounding the embryo. This process is especially important in seeds with physiological dormancy that is coat imposed, which requires the presence and activity of the seed coat and endosperm structures that form a barrier between the embryo and the external environment. In order to further understand the mechanisms by which the control of coat-induced dormancy takes place in Arabidopsis (Arabidopsis thaliana), Fedi et al. (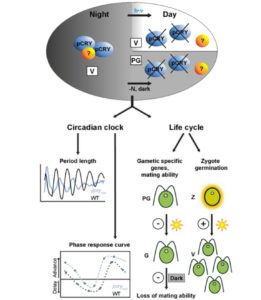 Cryptochromes are flavin-binding proteins that act as blue light receptors in bacteria, fungi, plants and insects and are components of the circadian oscillator in mammals. Animal and plant cryptochromes are evolutionarily divergent, although the unicellular alga Chlamydomonas reinhardtii has both an animal-like cryptochrome and a plant cryptochrome (pCRY; formerly CPH1). Müller et al. (
Cryptochromes are flavin-binding proteins that act as blue light receptors in bacteria, fungi, plants and insects and are components of the circadian oscillator in mammals. Animal and plant cryptochromes are evolutionarily divergent, although the unicellular alga Chlamydomonas reinhardtii has both an animal-like cryptochrome and a plant cryptochrome (pCRY; formerly CPH1). Müller et al. (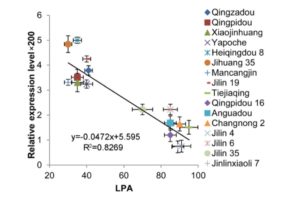 Soybean (Glycine max) is one of the most important oilseed crops that provides edible oil for humans and is a major renewable feedstock for biodiesel production around the world. As such, increasing soybean yield potential has become a long-term breeding objective. Soybean leaf petiole angle is an important plant architectural trait that affects canopy coverage, photosynthetic efficiency, and ultimately productivity in many legume crops. Leaf petiole angle is mainly controlled by the structure of the pulvinus at the base of a leaf petiole, leaf, or leaflet. Typically, a pulvinus consists of a core of vascular tissues surrounded by a flexible, bulky cylinder of thin-walled parenchyma cells. The outer cells of the parenchyma, termed the motor cells, are responsible for nyctinastic and thigmonastic movement through water-driven volume changes. Gao et al. (
Soybean (Glycine max) is one of the most important oilseed crops that provides edible oil for humans and is a major renewable feedstock for biodiesel production around the world. As such, increasing soybean yield potential has become a long-term breeding objective. Soybean leaf petiole angle is an important plant architectural trait that affects canopy coverage, photosynthetic efficiency, and ultimately productivity in many legume crops. Leaf petiole angle is mainly controlled by the structure of the pulvinus at the base of a leaf petiole, leaf, or leaflet. Typically, a pulvinus consists of a core of vascular tissues surrounded by a flexible, bulky cylinder of thin-walled parenchyma cells. The outer cells of the parenchyma, termed the motor cells, are responsible for nyctinastic and thigmonastic movement through water-driven volume changes. Gao et al. (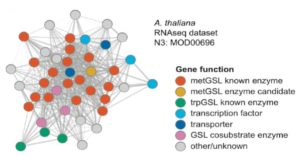 Plants produce scores of specialized metabolites (SMs) to attract or repel the organisms around them and to cope with life in a variable environment. For thousands of years, we have been exploiting these compounds to feed, heal, and adorn us. Many more SMs remain to be discovered: the chemical constituents of only 15% of the estimated 350,000 plant species on Earth have thus far been explored (
Plants produce scores of specialized metabolites (SMs) to attract or repel the organisms around them and to cope with life in a variable environment. For thousands of years, we have been exploiting these compounds to feed, heal, and adorn us. Many more SMs remain to be discovered: the chemical constituents of only 15% of the estimated 350,000 plant species on Earth have thus far been explored (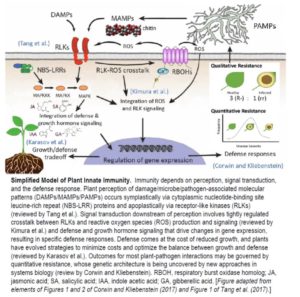 Tender green leaves and tasty tubers, roots, and stems are vulnerable to a wide range of pathogens, pests, and herbivores. Perhaps it should not be surprising that they have evolved an equally wide range of defense mechanisms. This issue of The Plant Cell includes reviews of just a few of the many facets of plant immunity.
Tender green leaves and tasty tubers, roots, and stems are vulnerable to a wide range of pathogens, pests, and herbivores. Perhaps it should not be surprising that they have evolved an equally wide range of defense mechanisms. This issue of The Plant Cell includes reviews of just a few of the many facets of plant immunity.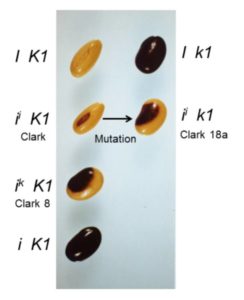 Most soybean seeds you see, whether in bins at the store, or in train cars as a commodity crop, have a yellow seed coat that may have only a tiny fleck of dark pigment at the hilum, where the seed attaches to the pod. The predominant yellow color results from silencing of chalcone synthase (CHS) genes by RNA interference (RNAi) due to the presence of I or ii alleles. The ii allele has an inverted duplication of CHS1, CHS3, and CHS4, causing the production of small interfering RNAs (siRNAs) that target CHS mRNAs and prevent pigment synthesis. Seeds carrying the rare ik allele have a larger region of pigment, producing a saddle pattern of dark purple pigment with black appearance on the yellow bean, more like a black-eyed pea. In a classic example of epistasis, the recessive k1 allele of the K1 locus overcomes the restriction of pigment to the hilum in ii plants: ii K1 soybeans have yellow seeds with a black hilum, but ii k1 soybeans have seeds with a black saddle (similar to ik K1 seeds), and I k1 soybeans have seeds that are entirely black.
Most soybean seeds you see, whether in bins at the store, or in train cars as a commodity crop, have a yellow seed coat that may have only a tiny fleck of dark pigment at the hilum, where the seed attaches to the pod. The predominant yellow color results from silencing of chalcone synthase (CHS) genes by RNA interference (RNAi) due to the presence of I or ii alleles. The ii allele has an inverted duplication of CHS1, CHS3, and CHS4, causing the production of small interfering RNAs (siRNAs) that target CHS mRNAs and prevent pigment synthesis. Seeds carrying the rare ik allele have a larger region of pigment, producing a saddle pattern of dark purple pigment with black appearance on the yellow bean, more like a black-eyed pea. In a classic example of epistasis, the recessive k1 allele of the K1 locus overcomes the restriction of pigment to the hilum in ii plants: ii K1 soybeans have yellow seeds with a black hilum, but ii k1 soybeans have seeds with a black saddle (similar to ik K1 seeds), and I k1 soybeans have seeds that are entirely black.