Verification of Arabidopsis stock collections using SNPmatch, a tool for genotyping high-plexed samples
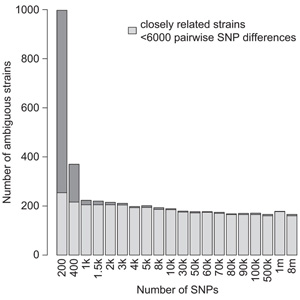 Experiments using large germplasm collections are prone to contamination. With dropping sequencing costs, it becomes easier to validate the genotype using minimal sequencing coverage. Pisupati and colleagues developed an open-source python pipeline, called SNPmatch, allowing identification of 930 Arabidopsis accessions from 1001 Genomes panel, based on 2,000 SNPs obtained by sequencing with minimal genome coverage. Differentiating between more closely related strains, differing by less than 6,000 SNPs, higher coverage was required. 74 mismatched lines were identified in 1001 Genomes panel. The user-friendly AraGeno web-application can be used by anyone to validate their own Arabidopsis stocks. The pipeline can be used on transcriptome or bisulfite sequencing data and has a potential to be extended to other plant species, allowing for quick and easy validation of germplasm.
Experiments using large germplasm collections are prone to contamination. With dropping sequencing costs, it becomes easier to validate the genotype using minimal sequencing coverage. Pisupati and colleagues developed an open-source python pipeline, called SNPmatch, allowing identification of 930 Arabidopsis accessions from 1001 Genomes panel, based on 2,000 SNPs obtained by sequencing with minimal genome coverage. Differentiating between more closely related strains, differing by less than 6,000 SNPs, higher coverage was required. 74 mismatched lines were identified in 1001 Genomes panel. The user-friendly AraGeno web-application can be used by anyone to validate their own Arabidopsis stocks. The pipeline can be used on transcriptome or bisulfite sequencing data and has a potential to be extended to other plant species, allowing for quick and easy validation of germplasm.
(Summary by Magdalena Julkowska) Sci. Data 10.1038/sdata.2017.184