What We’re Reading: April 20th
 This week’s issue of What We’re Reading is guest edited by Sunil Kumar Kenchanmane Raju, a postdoc in Chad Niederhuth’s lab at Michigan State University. He is interested in understanding the epigenetic and epigenomic diversity across land plants. Sunil is a postdoc ambassador of ASPB and social media coordinator for the CSHL plant course. This week, Sunil has selected papers with a focus on epigenetics and epigenomics. Enjoy!
This week’s issue of What We’re Reading is guest edited by Sunil Kumar Kenchanmane Raju, a postdoc in Chad Niederhuth’s lab at Michigan State University. He is interested in understanding the epigenetic and epigenomic diversity across land plants. Sunil is a postdoc ambassador of ASPB and social media coordinator for the CSHL plant course. This week, Sunil has selected papers with a focus on epigenetics and epigenomics. Enjoy!
A Kinesin-14 motor activates neocentromeres to promote meiotic drive in maize
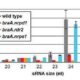
 Meiotic drive is essentially a subversion of meiosis such that particular regions or alleles are preferentially favored for transmission to the progeny. Abnormal chromosome 10 (Ab10) is a classic example of meiotic drive in maize that converts heterochromatic chromosomal knobs into motile ‘neocentromeres’. When Ab10 is present in the genome, all knobs move earlier and faster than centromeres, ultimately delivering knobbed chromatids into upper and lower cells of the tetrads during meiosis. This ensures preferential segregation of Ab10 and knobs to the progeny, as the lower cell goes on to form the egg. Dawe et al. used genomic approaches to identify a previously unknown region in Ab10 containing at least eight tandemly arrayed copies of Kinesin driver (Kindr). Two Ab10 mutants that lack meiotic drive turned out to be epimutants of kindr. These epimutants have increased DNA methylation directly over the promoter and small RNA accumulation on the promoter and first intron, resulting in decreases in Kindr gene expression. RNAi suppression of kindr showed the expected reduction in preferential transmission, proving its requirement in neocentromeric activity and meiotic drive. The authors show that KINDR belongs to the Kinesin-14A family, and at least in vitro, generates faster microtubule gliding capacity than its closest maize homolog ZmKIN11. Kindr and ZMKIN11 share ~94% amino acid identity, and phylogenetic analysis suggests a divergence time of ~11.8 MY. This period coincides with the divergence of maize and sorghum. Sorghum diverged from maize ~12 MYAa anddoes not have maize-like knobs, whereas Tripsacum dactyloides, which diverged from maize ~1 MYA, has knobs similar to that of maize. KINDR specifically binds to knobs with 180 bp repeats and not those containing TR1 repeats. Ab10 has a profound effect on maize, adding ~500 Mb of DNA to the genome and affecting segregation of a substantial number of genes. This potential of centromeric sequences to drive their preferential transmission may have led to epigenetic mechanisms to counter their inheritance. (Summary by Sunil Kumar Kenchanmane Raju) Cell 10.1016/j.cell.2018.03.009
Meiotic drive is essentially a subversion of meiosis such that particular regions or alleles are preferentially favored for transmission to the progeny. Abnormal chromosome 10 (Ab10) is a classic example of meiotic drive in maize that converts heterochromatic chromosomal knobs into motile ‘neocentromeres’. When Ab10 is present in the genome, all knobs move earlier and faster than centromeres, ultimately delivering knobbed chromatids into upper and lower cells of the tetrads during meiosis. This ensures preferential segregation of Ab10 and knobs to the progeny, as the lower cell goes on to form the egg. Dawe et al. used genomic approaches to identify a previously unknown region in Ab10 containing at least eight tandemly arrayed copies of Kinesin driver (Kindr). Two Ab10 mutants that lack meiotic drive turned out to be epimutants of kindr. These epimutants have increased DNA methylation directly over the promoter and small RNA accumulation on the promoter and first intron, resulting in decreases in Kindr gene expression. RNAi suppression of kindr showed the expected reduction in preferential transmission, proving its requirement in neocentromeric activity and meiotic drive. The authors show that KINDR belongs to the Kinesin-14A family, and at least in vitro, generates faster microtubule gliding capacity than its closest maize homolog ZmKIN11. Kindr and ZMKIN11 share ~94% amino acid identity, and phylogenetic analysis suggests a divergence time of ~11.8 MY. This period coincides with the divergence of maize and sorghum. Sorghum diverged from maize ~12 MYAa anddoes not have maize-like knobs, whereas Tripsacum dactyloides, which diverged from maize ~1 MYA, has knobs similar to that of maize. KINDR specifically binds to knobs with 180 bp repeats and not those containing TR1 repeats. Ab10 has a profound effect on maize, adding ~500 Mb of DNA to the genome and affecting segregation of a substantial number of genes. This potential of centromeric sequences to drive their preferential transmission may have led to epigenetic mechanisms to counter their inheritance. (Summary by Sunil Kumar Kenchanmane Raju) Cell 10.1016/j.cell.2018.03.009
Maternal components of RdDM are required for seed development in Brassica rapa
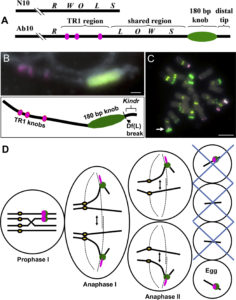
 RNA directed DNA methylation (RdDM) is an epigenetic process in which plant double-strand RNAs are processed into small RNAs (sRNA) that add repressive DNA methylation to homologous DNA sequences. RdDM primarily acts on repetitive DNA and transposable elements (TEs). Despite its important biological functions, loss of RdDM components in Arabidopsis does not show much phenotypic consequences. In contrast, mutations in RdDM components in genomes with higher transposons content like maize and tomato result in severe phenotypic consequences including reproductive defects. Grover et al. show that mutations in Pol IV-dependent sRNA pathway cause severe reproductive defects, specifically during seed production in Brassica rapa. High rates of abortion occur when the RdDM mutants are crossed as the female parent, and not when the mutants are used as pollen donors. No vegetative growth defects associated with loss of RdDM were observed in these mutants. However, mutant siliques were smaller and produced fewer viable seeds. 24nt siRNA, the abundant class of siRNAs in reproductive tissues, are strongly reduced in the B. rapa mutants. The authors show that seed production defect is due to loss of p4-siRNA activity in the maternal sporophytic tissue even though the defects manifest later during seed development. In conclusion, the authors suggest that the control of developing progeny by parental small RNA is a common biological mechanism and B. rapa, as a recently outbreeding species, may be invaluable in understanding the role of RdDM in plant reproduction. Plant J. 10.1111/tpj.13910
RNA directed DNA methylation (RdDM) is an epigenetic process in which plant double-strand RNAs are processed into small RNAs (sRNA) that add repressive DNA methylation to homologous DNA sequences. RdDM primarily acts on repetitive DNA and transposable elements (TEs). Despite its important biological functions, loss of RdDM components in Arabidopsis does not show much phenotypic consequences. In contrast, mutations in RdDM components in genomes with higher transposons content like maize and tomato result in severe phenotypic consequences including reproductive defects. Grover et al. show that mutations in Pol IV-dependent sRNA pathway cause severe reproductive defects, specifically during seed production in Brassica rapa. High rates of abortion occur when the RdDM mutants are crossed as the female parent, and not when the mutants are used as pollen donors. No vegetative growth defects associated with loss of RdDM were observed in these mutants. However, mutant siliques were smaller and produced fewer viable seeds. 24nt siRNA, the abundant class of siRNAs in reproductive tissues, are strongly reduced in the B. rapa mutants. The authors show that seed production defect is due to loss of p4-siRNA activity in the maternal sporophytic tissue even though the defects manifest later during seed development. In conclusion, the authors suggest that the control of developing progeny by parental small RNA is a common biological mechanism and B. rapa, as a recently outbreeding species, may be invaluable in understanding the role of RdDM in plant reproduction. Plant J. 10.1111/tpj.13910
DNA methylation dynamics during early plant life
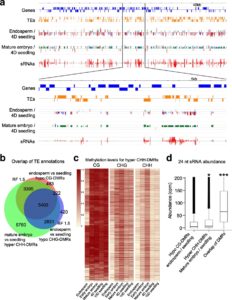 DNA methylation is extensively reprogrammed in the early embryo and germlines of mammals, whereas flowering plants do not show such extensive resetting except in the endosperm. Active DNA demethylation in the central cell and reduced activity of DNA methyltransferases leads to global hypomethylation of the maternal genome in the endosperm. On the paternal side, the vegetative nucleus of the pollen undergoes active DNA demethylation, but this is limited to only a few transposable element (TE) sequences. Bouyer et al. used Arabidopsis to examine DNA methylation dynamics during embryogenesis and early vegetative development. They show higher non-CG methylation in the mature embryo compared to seedlings and adult plants. Methylation at CHH sites (mCHH) makes up 44% of all methylated cytosines in embryos, compared to less than 27% in seedlings, particularly in the pericentromeric regions. CHH hypermethylation in mature embryo coincides with the hypomethylated state during endosperm development and a higher abundance of 24nt siRNAs. Based on their analysis of temporal gene expression and patterns of mCHH on long TE sequences in the early and mature embryo, the authors suggest that siRNA production in the endosperm precedes RNA-directed DNA Methylation (RdDM) in the mature embryo. RdDM is active in every cell of the mature embryo in contrast to its activity being restricted to meristems post-embryonically. They also show that failure to terminate the embryonic developmental program as seen in fie mutants leads to massive CHH hypermethylation retained in the seedlings, particularly on RdDM targets. (Summary by Sunil Kumar) Genome Biology 10.1186/s13059-017-1313-0
DNA methylation is extensively reprogrammed in the early embryo and germlines of mammals, whereas flowering plants do not show such extensive resetting except in the endosperm. Active DNA demethylation in the central cell and reduced activity of DNA methyltransferases leads to global hypomethylation of the maternal genome in the endosperm. On the paternal side, the vegetative nucleus of the pollen undergoes active DNA demethylation, but this is limited to only a few transposable element (TE) sequences. Bouyer et al. used Arabidopsis to examine DNA methylation dynamics during embryogenesis and early vegetative development. They show higher non-CG methylation in the mature embryo compared to seedlings and adult plants. Methylation at CHH sites (mCHH) makes up 44% of all methylated cytosines in embryos, compared to less than 27% in seedlings, particularly in the pericentromeric regions. CHH hypermethylation in mature embryo coincides with the hypomethylated state during endosperm development and a higher abundance of 24nt siRNAs. Based on their analysis of temporal gene expression and patterns of mCHH on long TE sequences in the early and mature embryo, the authors suggest that siRNA production in the endosperm precedes RNA-directed DNA Methylation (RdDM) in the mature embryo. RdDM is active in every cell of the mature embryo in contrast to its activity being restricted to meristems post-embryonically. They also show that failure to terminate the embryonic developmental program as seen in fie mutants leads to massive CHH hypermethylation retained in the seedlings, particularly on RdDM targets. (Summary by Sunil Kumar) Genome Biology 10.1186/s13059-017-1313-0
Extensive transcriptomic and epigenomic remodelling occurs during Arabidopsis thaliana germination
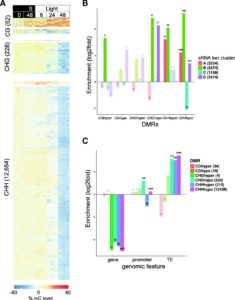 Understanding the complex regulatory mechanisms that contribute to germination and seedling establishment requires integration of gene-expression, transcription factors (TFs), DNA methylation, smallRNA data, and their interactions. Narsai et al. describe the first dynamic transcription factor network model of Arabidopsis seed germination over an extensive time-course, showing widespread alternative splicing and changes in known and novel regulatory genes. Their results uncovered both known germination regulators and novel TFs that control specific stages of germination using Dynamic Regulatory Events Miner (DREM) modeling of time course transcriptome data. The authors validate their results by showing delayed germination in T-DNA insertion lines carrying mutations in transcription factor genes identified using DREM. For the majority of these TFs, their ability to bind DNA is negatively affected by DNA methylation, shaping the regulatory network during vegetative growth. The authors also show significant changes in both miRNA and siRNA expression during seed germination and the transition to seedlings. They speculate whether miRNAs mediate DNA methylation changes by targeting genes involved in DNA methylation like DRM2 and IDNL-2. The epigenome undertoes extensive demethylation during the transition from embryo to seedling and there is significant overlap between CHH-hypomethylated DMRs and siRNA clusters, indicating that the decrease in CHH methylation is likely due to the RNA-directed DNA Methylation pathway (RdDM) and not the CMT2-mediated pathway. Genome Biology 10.1186/s13059-017-1302-3
Understanding the complex regulatory mechanisms that contribute to germination and seedling establishment requires integration of gene-expression, transcription factors (TFs), DNA methylation, smallRNA data, and their interactions. Narsai et al. describe the first dynamic transcription factor network model of Arabidopsis seed germination over an extensive time-course, showing widespread alternative splicing and changes in known and novel regulatory genes. Their results uncovered both known germination regulators and novel TFs that control specific stages of germination using Dynamic Regulatory Events Miner (DREM) modeling of time course transcriptome data. The authors validate their results by showing delayed germination in T-DNA insertion lines carrying mutations in transcription factor genes identified using DREM. For the majority of these TFs, their ability to bind DNA is negatively affected by DNA methylation, shaping the regulatory network during vegetative growth. The authors also show significant changes in both miRNA and siRNA expression during seed germination and the transition to seedlings. They speculate whether miRNAs mediate DNA methylation changes by targeting genes involved in DNA methylation like DRM2 and IDNL-2. The epigenome undertoes extensive demethylation during the transition from embryo to seedling and there is significant overlap between CHH-hypomethylated DMRs and siRNA clusters, indicating that the decrease in CHH methylation is likely due to the RNA-directed DNA Methylation pathway (RdDM) and not the CMT2-mediated pathway. Genome Biology 10.1186/s13059-017-1302-3
Similarity between soybean and Arabidopsis seed methylomes and loss of non-CG methylation does not affect seed development
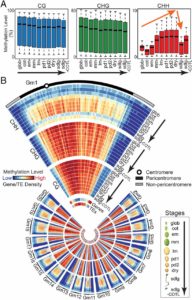 Different parts of the seed have distinct genetic origins and functions. The seed coat is maternally derived from ovule integuments, whereas the embryo descends from the fertilized egg and the endosperm from the central cell. Lin et al. profiled the methylome landscape during seed development and germination in Arabidopsis and soybean using laser capture microdissection and whole-genome bisulfite sequencing. DNA methylation architecture during seed development and germination appears to be conserved between soybean and Arabidopsis despite ~90 MY divergence. They show that CHH methylation drastically increases during seed development, targeting all classes of transposable elements (TEs). Later during germination, both the bulk CHH methylation levels and the number of mCHH sites decreases. In contrast, CG and CHG methylation levels remain more or less constant during seed development. To understand the role of DNA methylation changes on the expression of genes known to be important for seed development and germination in both soybean and Arabidopsis, the authors characterized the methylation patterns and expression levels of ~75 genes. Most of these genes were found in regions of very low DNA methylation, which the authors termed ‘demethylated valleys’. The surrounding methylation levels of these genes didn’t change during seed development, despite changes in gene expression. The authors suggest that the regulation of seed development genes results primarily from transcriptional events that are independent of DNA methylation changes. Furthermore, Arabidopsis mutants lacking CHG and CHH methylation develop and germinate normally and don’t show much change to the transcriptome, except for the increased expression of transposons. This suggests that CHH hypermethylation during seed development may be a failsafe mechanism to avoid deleterious effects of TE proliferation. PNAS
Different parts of the seed have distinct genetic origins and functions. The seed coat is maternally derived from ovule integuments, whereas the embryo descends from the fertilized egg and the endosperm from the central cell. Lin et al. profiled the methylome landscape during seed development and germination in Arabidopsis and soybean using laser capture microdissection and whole-genome bisulfite sequencing. DNA methylation architecture during seed development and germination appears to be conserved between soybean and Arabidopsis despite ~90 MY divergence. They show that CHH methylation drastically increases during seed development, targeting all classes of transposable elements (TEs). Later during germination, both the bulk CHH methylation levels and the number of mCHH sites decreases. In contrast, CG and CHG methylation levels remain more or less constant during seed development. To understand the role of DNA methylation changes on the expression of genes known to be important for seed development and germination in both soybean and Arabidopsis, the authors characterized the methylation patterns and expression levels of ~75 genes. Most of these genes were found in regions of very low DNA methylation, which the authors termed ‘demethylated valleys’. The surrounding methylation levels of these genes didn’t change during seed development, despite changes in gene expression. The authors suggest that the regulation of seed development genes results primarily from transcriptional events that are independent of DNA methylation changes. Furthermore, Arabidopsis mutants lacking CHG and CHH methylation develop and germinate normally and don’t show much change to the transcriptome, except for the increased expression of transposons. This suggests that CHH hypermethylation during seed development may be a failsafe mechanism to avoid deleterious effects of TE proliferation. PNAS
Loss of mCHH islands in maize chromomethylase and DDM1-type nucleosome remodeler mutants
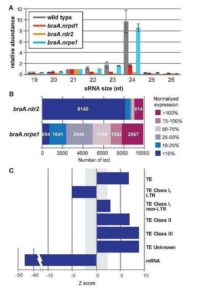
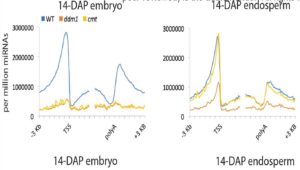 Plant DNA methylation in different sequence contexts is catalyzed by distinct types of methyltransferases. METHYLTRANSFERASE 1 (MET1) is responsible for CG methylation, CHROMOMETHYLASE (CMT1, 2, and 3) methylates CHG and CHH, while DOMAINS REARRANGED METHYLTRANSFERASE (DRM1 and 2) methylates in all three contexts through the RNA-directed DNA Methylation (RdDM) pathway. DECREASED DNA METHYLATION1 (DDM1) provides DNA methyltransferase access to heterochromatin to silence transposable elements together with the RdDM pathway. Single mutants of CMT (ZMET2 and ZMET5) and DDM1 (CHR101 and CHR106) in maize show some defects in DNA methylation, while double mutants of chr101 and chr106 and double mutants of zmet2 and zmet5 are nonviable. Arabidopsis mutants of the equivalent genes produce DNA methylation and small RNA changes, but little or no phenotypic consequences at the whole plant level. Fu et al. attempt to understand the relationship between RdDM and other forms of DNA methylation by looking at methylation and small RNA signatures produced by double knockouts of CMT (ZMET2 and ZMET5) and DDM1 (CHR101 and CHR106) in maize. The authors show that double mutants produce embryo and endosperm, and examine DNA methylation and small RNA changes in both tissues. In the developing embryo, there was a complete loss of 24nt siRNA and CHH methylation at CHH islands. 21nt and 22nt siRNA were gained in heterochromatin, although these 21-22nt siRNAs did not direct DNA methylation of the heterochromatic loci. In both double mutants, the dominant siRNA type shifted to 21 and 22nt, while 24nt siRNAs from terminal inverted repeat transposons were reduced eight-fold. The ddm1 and cmt mutants’ stronger effect on RdDM in maize may be attributed to its larger genome size compared to Arabidopsis. bioRxiv
Plant DNA methylation in different sequence contexts is catalyzed by distinct types of methyltransferases. METHYLTRANSFERASE 1 (MET1) is responsible for CG methylation, CHROMOMETHYLASE (CMT1, 2, and 3) methylates CHG and CHH, while DOMAINS REARRANGED METHYLTRANSFERASE (DRM1 and 2) methylates in all three contexts through the RNA-directed DNA Methylation (RdDM) pathway. DECREASED DNA METHYLATION1 (DDM1) provides DNA methyltransferase access to heterochromatin to silence transposable elements together with the RdDM pathway. Single mutants of CMT (ZMET2 and ZMET5) and DDM1 (CHR101 and CHR106) in maize show some defects in DNA methylation, while double mutants of chr101 and chr106 and double mutants of zmet2 and zmet5 are nonviable. Arabidopsis mutants of the equivalent genes produce DNA methylation and small RNA changes, but little or no phenotypic consequences at the whole plant level. Fu et al. attempt to understand the relationship between RdDM and other forms of DNA methylation by looking at methylation and small RNA signatures produced by double knockouts of CMT (ZMET2 and ZMET5) and DDM1 (CHR101 and CHR106) in maize. The authors show that double mutants produce embryo and endosperm, and examine DNA methylation and small RNA changes in both tissues. In the developing embryo, there was a complete loss of 24nt siRNA and CHH methylation at CHH islands. 21nt and 22nt siRNA were gained in heterochromatin, although these 21-22nt siRNAs did not direct DNA methylation of the heterochromatic loci. In both double mutants, the dominant siRNA type shifted to 21 and 22nt, while 24nt siRNAs from terminal inverted repeat transposons were reduced eight-fold. The ddm1 and cmt mutants’ stronger effect on RdDM in maize may be attributed to its larger genome size compared to Arabidopsis. bioRxiv
Comparison of the relative potential for epigenetic and genetic variation to contribute to trait stability
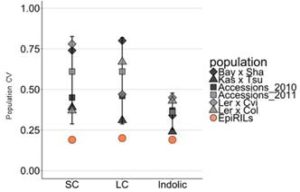 Epigenetic variation, both natural and induced, can influence heritable phenotypic variation of complex traits. Understanding the effect of epigenetic changes on trait variation is complicated by the confounding effects of DNA sequence polymorphisms. To overcome this complication, epigenetic recombinant inbred lines (epiRILs) have been developed from parental lines with little or no DNA sequence variation, but differing in DNA methylation patterns. Aller et al. used Arabidopsis epiRILs to compare the potential of epigenetic and genetic variation to influence adaptive traits. Specifically, they look at the correlation between DNA methylation changes and accumulation of glucosinolates, defense compounds produced by almost all plants of the order Brassicales. Potential epigenetic heritability (epi-heritability) of glucosinolate variation ranged from 10% – 13% in the epiRILS, much lower than estimates of 30% to 80% heritability in genetically diverse populations. Also, the authors found that glucosinolate epi-heritability was lower than the epi-heritability for flowering time (32%). The authors then identified epiQTLs containing differentially methylated regions (DMRs) correlating with variance in glucosinolate levels. They did not find any significant epiQTL overlapping known genes controlling glucosinolates, perhaps indicating that the DNA methylation differences affect loci with unknown causal effect or a different suite of genes with small effects on the trait. They also found DMRs linked to both increased and decreased glucosinolate levels compared to wild type. In contrast, for flowering time epiQTL, these were all skewed towards earlier flowering, highlighting fundamental differences in how DNA methylation variation influences different traits. For example, DNA methylation may act as an irreversible switch for traits like flowering, which cannot stop once the process is initiated; while for other traits like biosynthesis, genes in different steps can be affected to increase or decrease production, accumulation, transport etc. They conclude that the contribution of epigenetic variation to glucosinolate variation is much lower than the effect of genetic variation. They also suggest that DNA methylation changes may have an effect on trait stability for both glucosinolates and flowering time. G3
Epigenetic variation, both natural and induced, can influence heritable phenotypic variation of complex traits. Understanding the effect of epigenetic changes on trait variation is complicated by the confounding effects of DNA sequence polymorphisms. To overcome this complication, epigenetic recombinant inbred lines (epiRILs) have been developed from parental lines with little or no DNA sequence variation, but differing in DNA methylation patterns. Aller et al. used Arabidopsis epiRILs to compare the potential of epigenetic and genetic variation to influence adaptive traits. Specifically, they look at the correlation between DNA methylation changes and accumulation of glucosinolates, defense compounds produced by almost all plants of the order Brassicales. Potential epigenetic heritability (epi-heritability) of glucosinolate variation ranged from 10% – 13% in the epiRILS, much lower than estimates of 30% to 80% heritability in genetically diverse populations. Also, the authors found that glucosinolate epi-heritability was lower than the epi-heritability for flowering time (32%). The authors then identified epiQTLs containing differentially methylated regions (DMRs) correlating with variance in glucosinolate levels. They did not find any significant epiQTL overlapping known genes controlling glucosinolates, perhaps indicating that the DNA methylation differences affect loci with unknown causal effect or a different suite of genes with small effects on the trait. They also found DMRs linked to both increased and decreased glucosinolate levels compared to wild type. In contrast, for flowering time epiQTL, these were all skewed towards earlier flowering, highlighting fundamental differences in how DNA methylation variation influences different traits. For example, DNA methylation may act as an irreversible switch for traits like flowering, which cannot stop once the process is initiated; while for other traits like biosynthesis, genes in different steps can be affected to increase or decrease production, accumulation, transport etc. They conclude that the contribution of epigenetic variation to glucosinolate variation is much lower than the effect of genetic variation. They also suggest that DNA methylation changes may have an effect on trait stability for both glucosinolates and flowering time. G3
TET-mediated epimutagenesis of the Arabidopsis thaliana methylome
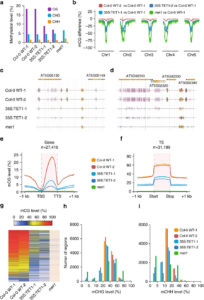 Natural and induced DNA methylation variations are known to alter gene expression changes that may ultimately be important for agronomically important traits. Epigenetically manipulating plant methylomes to create heritable changes can be invaluable in crop improvement programs. Ji et al. describe the expression of a human ten eleven translocation enzyme (TET) in Arabidopsis thaliana to create random demethylation of the genome. The widespread hypomethylation mimics mutants lacking the MET1 DNA methyltransferase, although with less severe phenotypes. hTET1cd expression in Arabidopsis resulted in a global reduction in CG methylation, while its effect was not as severe on CHG and CHH methylation. Individual transgenic lines showed varying levels of reduction in CG methylation, making it a suitable system to select for desired levels of demethylation. Similar to met1, hTET1 plants showed hypomethylation of the seventh intron of INCREASE IN BONSAI METHYLATION 1 (IBM1), a histone H3K9 demethylase, resulting in altered expression and CHG hypermethylation in gene body methylated (gBM) loci. Similarly, they also show loss of methylation at FLOWERING WAGENINGEN (FWA), associated with its increased expression and a delayed flowering phenotype. The authors also show that using the ACTIN 2 promoter (ACT2:sfGFP-hTET1cd) that has activity in all tissue types of juvenile plants substantially delays flowering time in the transgenic plants compared to lines with 35S:hTET1cd. The late flowering phenotype was stably inherited in the progeny irrespective of the presence of transgene. In contrast, in the 35S:hTET1cd, the transgenerational inheritance was minimum after removal of the transgene. TET-mediated engineering of DNA methylation states to create epialleles that are faithfully inherited in subsequent generations in agriculturally important crop species is an interesting area for further investigation. Nature Comms. 10.1038/s41467-018-03289-7
Natural and induced DNA methylation variations are known to alter gene expression changes that may ultimately be important for agronomically important traits. Epigenetically manipulating plant methylomes to create heritable changes can be invaluable in crop improvement programs. Ji et al. describe the expression of a human ten eleven translocation enzyme (TET) in Arabidopsis thaliana to create random demethylation of the genome. The widespread hypomethylation mimics mutants lacking the MET1 DNA methyltransferase, although with less severe phenotypes. hTET1cd expression in Arabidopsis resulted in a global reduction in CG methylation, while its effect was not as severe on CHG and CHH methylation. Individual transgenic lines showed varying levels of reduction in CG methylation, making it a suitable system to select for desired levels of demethylation. Similar to met1, hTET1 plants showed hypomethylation of the seventh intron of INCREASE IN BONSAI METHYLATION 1 (IBM1), a histone H3K9 demethylase, resulting in altered expression and CHG hypermethylation in gene body methylated (gBM) loci. Similarly, they also show loss of methylation at FLOWERING WAGENINGEN (FWA), associated with its increased expression and a delayed flowering phenotype. The authors also show that using the ACTIN 2 promoter (ACT2:sfGFP-hTET1cd) that has activity in all tissue types of juvenile plants substantially delays flowering time in the transgenic plants compared to lines with 35S:hTET1cd. The late flowering phenotype was stably inherited in the progeny irrespective of the presence of transgene. In contrast, in the 35S:hTET1cd, the transgenerational inheritance was minimum after removal of the transgene. TET-mediated engineering of DNA methylation states to create epialleles that are faithfully inherited in subsequent generations in agriculturally important crop species is an interesting area for further investigation. Nature Comms. 10.1038/s41467-018-03289-7