In the Transcripts: Long-read Transcriptomics Enables a Novel Type of Transposable Element Annotation in Plants
Transposable Elements (TEs) are mobile genetic elements and major constituents of eukaryotic chromosomes. TEs promote genetic and epigenetic variation within genomes and are a major source of evolutionary novelty and adaptation (Lisch, 2013). In plants, TEs represent from 20% of the genomic content in Arabidopsis thaliana to 85% in maize (Zea mays). Structural characterization of TEs and identification of transcriptionally active copies is critical to understand how TEs alter gene expression, chromatin topology and cellular and organismal growth, development and response to the environment. Compared to genes, the repetitiveness and structural complexity of TEs have thus far restricted their annotation to minimal features and hampered their analysis. Notably, annotation of transcriptionally active elements and identification of transcript features, such as the transcription start site (TSS) and splicing patterns, are still lacking.
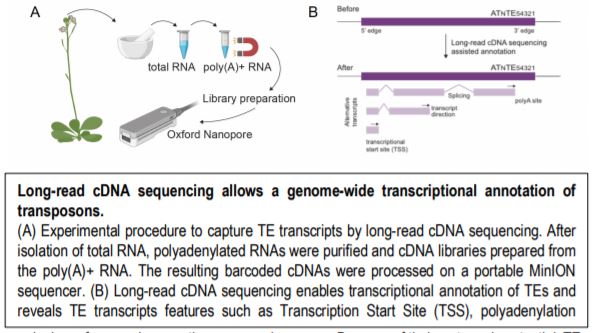 In a new study, Panda and Slotkin used Oxford Nanopore Technology long-read sequencing of cDNAs to establish a transcript-based annotation of TEs in Arabidopsis thaliana (Panda and Slotkin, 2020) (Figure 1A). To expose TE transcripts for sequencing and annotation, the authors used a combination of Arabidopsis mutants compromised in multiple layers of TE silencing. After generating over 5 million reads, the authors used the Oxford Nanopore Pinfish pipeline to annotate TE transcripts and identified a total of 2,188 TE transcript models originating from 1,292 individual TEs. The TE transcript models include newly identified TSSs, polyadenylation sites, transcription orientation and splicing patterns (Figure 1B). When overlaying these new TE transcript features onto the community-standard Arabidopsis TAIR10 TE annotation, the authors found that only 4% of previously annotated TEs can produce transcripts in the conditions tested, a large majority being Gypsy-like LTR retrotransposons and Mutator DNA transposons.
In a new study, Panda and Slotkin used Oxford Nanopore Technology long-read sequencing of cDNAs to establish a transcript-based annotation of TEs in Arabidopsis thaliana (Panda and Slotkin, 2020) (Figure 1A). To expose TE transcripts for sequencing and annotation, the authors used a combination of Arabidopsis mutants compromised in multiple layers of TE silencing. After generating over 5 million reads, the authors used the Oxford Nanopore Pinfish pipeline to annotate TE transcripts and identified a total of 2,188 TE transcript models originating from 1,292 individual TEs. The TE transcript models include newly identified TSSs, polyadenylation sites, transcription orientation and splicing patterns (Figure 1B). When overlaying these new TE transcript features onto the community-standard Arabidopsis TAIR10 TE annotation, the authors found that only 4% of previously annotated TEs can produce transcripts in the conditions tested, a large majority being Gypsy-like LTR retrotransposons and Mutator DNA transposons.
A major issue in performing genome-wide TE analyses is the ambiguous mapping of reads to individual TE copies belonging to the same family. This is mostly due to the short read size of second-generation sequencing technologies that cannot discriminate between identical or near-identical TEs and results in multi-mapping reads that limit the resolution of the analysis. Using the multi-copy Evadé retrotransposon as an example, Panda and Slotkin demonstrated that short-read RNA-seq data fails at accurately identifying transcriptionally active Evadé copies due to identical multi-mapping reads. The same analysis using the Oxford Nanopore Technology-generated long-reads accurately pinpointed the Evadé copies producing transcripts. In addition, using the transcript-based TE annotation as a guide for distributing the multi-mapping short reads dramatically enhanced the mapping specificity and reads were faithfully assigned to the corresponding transcribed Evadé copy. The authors further expanded their analysis to maize to validate the utility of such an approach in a larger and more complex plant genome. They took advantage of publicly available PacBio Iso-Seq data obtained from a diverse set of maize tissues, including endosperm and pollen that display developmental relaxation of TE silencing (Wang et al., 2016; Martínez and Slotkin, 2012). They detected over a thousand TE transcripts originating from 745 unique TEs, mostly shared between tissues but including a subset of pollen-specific TE transcripts. Similar to Arabidopsis Evadé, they identified element-specific transcripts from the high-copy (over 12,000) maize Opie transposons.
Because of their mutagenic potential, TEs are usually maintained in a repressed state by DNA methylation. In plants, TEs are decorated with peaks of CHH (in which H is any base other than G) methylation at their 5’ edges that insulates the TE from the surrounding genomic environment (Zemach et al., 2013). Panda and Slotkin applied their transcript-based annotation strategy to look at the prevalence of 5’ edge CHH peaks in transcriptionally potent transposons. They observed that TEs identified through transcript-based annotation exhibit higher CHH methylation at their 5’ edge than non-transcribed TEs, suggesting efficient targeting and silencing of these mutagenic elements by the genome. In addition to DNA methylation, TEs are silenced through the production of small interfering RNAs (siRNAs). The molecular mechanism by which plant TE transcripts are processed to generate primary siRNAs is still obscure. In other systems, TE transcripts display reduced splicing accuracy compared to genes and this perturbed splicing is thought to fuel the production of primary siRNAs. The authors thus investigated the splicing features of transcribed TEs identified in their analysis. They found that transcribed TEs have generally larger exons, fewer introns, and reduced splicing accuracy compared to protein-coding genes. However, in Arabidopsis, splicing accuracy does not correlate with the propensity of a TE transcript to generate primary siRNAs.
This long-read transcript-based transposon annotation represents a valuable resource for the plant community and enables genome-wide analyses and family-specific studies on TEs that have so far been hampered by the use of short-read sequencing approaches. Refinements of long-read single-molecule sequencing technologies offer an unprecedented opportunity to study transposon activities and their molecular control at scale in notoriously complex plant genomes (Shahid and Slotkin, 2020).
Matthias Benoit
Howard Hughes Medical Institute
Cold Spring Harbor Laboratory
ORCID: 0000-0002-3958-3173
REFERENCES
Lisch, D. (2013). How important are transposons for plant evolution? Nat. Rev. Genet. 14: 49–61.
Martínez, G. and Slotkin, R.K. (2012). Developmental relaxation of transposable element silencing in plants: functional or byproduct? Curr. Opin. Plant Biol. 15: 496–502.
Shahid, S. and Slotkin, R.K. (2020). The current revolution in transposable element biology enabled by long reads. Curr. Opin. Plant Biol. 54: 49–56.
Wang, B., Tseng, E., Regulski, M., Clark, T.A., Hon, T., Jiao, Y., Lu, Z., Olson, A., Stein, J.C., and Ware, D. (2016). Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 7: 11708.
Zemach, A., Kim, M.Y., Hsieh, P.-H., Coleman-Derr, D., Eshed-Williams, L., Thao, K., Harmer, S.L., and Zilberman, D. (2013). The Arabidopsis Nucleosome Remodeler DDM1 Allows DNA Methyltransferases to Access H1-Containing Heterochromatin. Cell 153: 193–205.
Panda, K. and Slotkin, R.K. (2020). Long-read cDNA Sequencing Enables a “Gene-Like” Transcript Annotation of Transposable Elements. The Plant Cell. https://doi.org/10.1105/tpc.20.00115