The Shifting Transcriptional Response of Corn Smut Fungus
As a biotrophic fungus, Ustilago maydis (corn smut fungus) relies on living plant tissues for sustenance. Once U. maydis cells of compatible mating types fuse on a leaf surface, they produce a dikaryotic filament with a specialized infection structure—the appressorium—that penetrates epidermal cells. Initially totally encased by the plant’s plasma membrane, the fungus then grows into the mesophyll and nutrient-rich vascular tissue of its host. The infected plant cells divide rapidly and enlarge, forming tumors that are filled with fungal hyphae and, with time, fungal spores (Lanver et al., 2017).
Colonization is possible due to the large arsenal of secreted effector proteins produced by the fungus, which subvert the plant’s defense responses and redirect the host´s metabolism to support fungal growth. Many of the 476 effectors secreted by U. maydis lack established structural or functional domains (Schuster et al., 2017), and only 5 of these have been characterized in detail (Lanver et al., 2017). Characterizing the remaining effectors and pinpointing the stages of infection at which they are active would provide insight into the biotrophic life cycle of this fungus, and possibly suggest ways to contain it.
Lanver et al. (2018) analyzed the U. maydis transcriptome at each of the plant-associated stages of its life cycle in maize (Zea mays), including the very early ones, when fungal biomass is low. Tissues of maize seedlings infected with compatible U. maydis wild-type strains were sampled at seven time points, from pre-penetration (0.5 days post infection) to mature spore formation (12 days post infection). mRNA libraries produced from samples taken at each time point were subjected to Illumina sequencing. After the maize genome was filtered from the >2 billion paired end reads obtained, the remaining reads were mapped to the U. maydis genome.
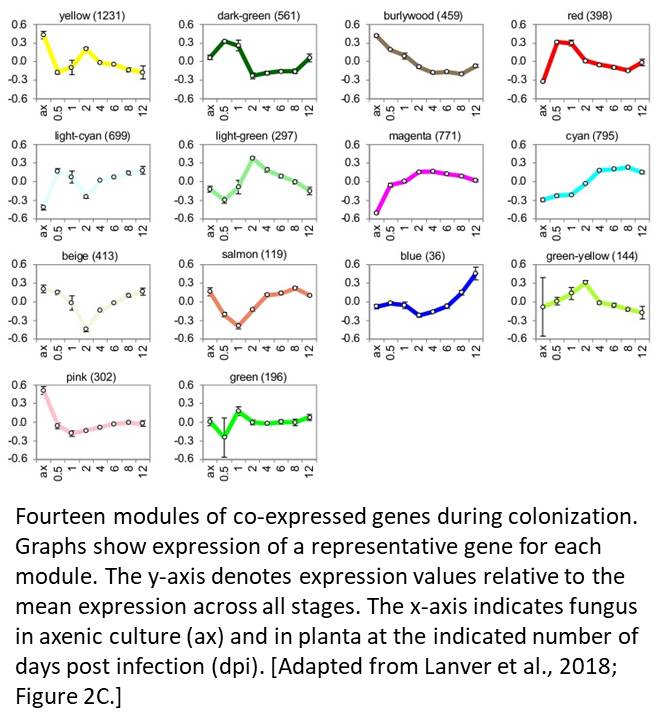
The authors found that 4586 genes (representing 68% of the U. maydis genome) were differentially expressed as pathogenesis progressed. By conducting a weighted gene co-expression network analysis, the team identified 14 modules (see figure), with 36 to 1231 genes each, that exhibited discrete waves of expression, some of which coincided with specific stages of fungal development. Using gene ontology analysis, the authors functionally annotated each of the modules, identifying key processes in each module. An in-depth analysis of the modules provided intriguing insights into defense suppression, tumor induction, and fungal nutrition.
In addition to revealing key changes in the fungal transcriptome associated with virulence, the data collected here represent a valuable resource for future studies aimed at deciphering the mechanisms used by this model fungus to colonize plants.
References
Lanver, D., Tollot, M., Schweizer, G., Lo Presti, L., Reissmann, S., Ma, L.S., Schuster, M., Tanaka, S., Liang, L., Ludwig, N., and Kahmann, R. (2017). Ustilago maydis effectors and their impact on virulence. Nat Rev Microbiol. doi: 10.1038/nrmicro.2017.33.
Lanver, D., Müller, A.N., Happel, P., Schweizer, G., Haas, F.B., Franitza, M., Pellegrin, C., Reissmann, S., Altmüller, J., Rensing, S.A., and Kahmann, R. (2018). The Biotrophic Development of Ustilago maydis Studied by RNAseq Analysis. Plant Cell: doi.org/10.1105/tpc.17.00764.
Schuster, M., Schweizer, G., and Kahmann, R. (2017). Comparative Analyses of Secreted Proteins in Plant Pathogenic Smut Fungi and Related Basidiomycetes. Fungal Genetics and Biology: https://doi.org/10.1016/j.fgb.2016.12.003.