Plant science research weekly: November 29
Review: Design and engineering of advanced plant optogenetics systems
 Optogenetics uses light-sensitive proteins to trigger specific outputs in the cell in response to particular wavelengths of light. Many optogenetic systems use chimeric proteins that contain different sensor and effector domains; with this, it is possible to use light to modulate DNA-binding activity, gene expression, or protein localization. An example is the use of PhyB-PIF3 in combination with GAL4 to generate a Red Light (RL)-driven promoter system. Additionally, proteins can be activated or their dimerization can be triggered in response to light. The use of these systems has excellent advantages, such as high resolution in time and space, quick responses, non-invasive delivery of the signal, and the potential for reversibility. In this review, Banerjee and Mitra provide an overview of key aspects to consider in the design and generation of optogenetics systems in plants. One of the first things to do is choose the chromophore and consider its bioavailability in the cell. Another feature to consider is how efficiently a photoreceptor converts excitation light into photochemical reactions (quantum efficiency), the intensity of light necessary to trigger responses, and the (de)activation dynamics. The authors highlight important design principles in representative examples of optogenetics systems, and discuss how the use of structural information, directed evolution, modeling, and in silico approaches can improve optogenetics systems. (Summary by Humberto Herrera-Ubaldo) Trends Plant Sci. 10.1016/j.tplants.2019.10.002
Optogenetics uses light-sensitive proteins to trigger specific outputs in the cell in response to particular wavelengths of light. Many optogenetic systems use chimeric proteins that contain different sensor and effector domains; with this, it is possible to use light to modulate DNA-binding activity, gene expression, or protein localization. An example is the use of PhyB-PIF3 in combination with GAL4 to generate a Red Light (RL)-driven promoter system. Additionally, proteins can be activated or their dimerization can be triggered in response to light. The use of these systems has excellent advantages, such as high resolution in time and space, quick responses, non-invasive delivery of the signal, and the potential for reversibility. In this review, Banerjee and Mitra provide an overview of key aspects to consider in the design and generation of optogenetics systems in plants. One of the first things to do is choose the chromophore and consider its bioavailability in the cell. Another feature to consider is how efficiently a photoreceptor converts excitation light into photochemical reactions (quantum efficiency), the intensity of light necessary to trigger responses, and the (de)activation dynamics. The authors highlight important design principles in representative examples of optogenetics systems, and discuss how the use of structural information, directed evolution, modeling, and in silico approaches can improve optogenetics systems. (Summary by Humberto Herrera-Ubaldo) Trends Plant Sci. 10.1016/j.tplants.2019.10.002
Comment: Climate tipping points — too risky to bet against
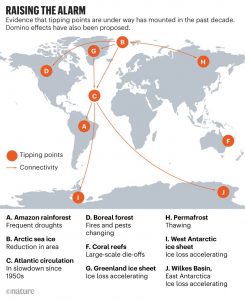 This Comment in Nature by Lenton et al. (leaders in climate study and policy) is making headlines, as it points to the serious implications of continuing climate change. It’s worth looking at the article itself, not just the digested version. The authors’ key message is that all the different places we’re seeing evidence for climate change are linked, and that these linkages are leading to faster adverse consequences than previously expected, as revealed by new data and models. As an example, melting Artic and Greenland ice causes freshwater influx into the North Atlantic (specifically, the Atlantic Meridional Overturning Circulation: AMOC) , changing rainfall patterns in Europe and Africa, as well as affecting the Amazon, East Asia, the Southern Ocean, and ultimately accelerating Antarctic ice loss. They also define an equation to calculate the magnitude of the emergency we are facing: E = R × U = p × D × τ / T, which is an interesting way to quantify how great the emergency we face is. (Summary by Mary Williams) Nature 10.1038/d41586-019-03595-0
This Comment in Nature by Lenton et al. (leaders in climate study and policy) is making headlines, as it points to the serious implications of continuing climate change. It’s worth looking at the article itself, not just the digested version. The authors’ key message is that all the different places we’re seeing evidence for climate change are linked, and that these linkages are leading to faster adverse consequences than previously expected, as revealed by new data and models. As an example, melting Artic and Greenland ice causes freshwater influx into the North Atlantic (specifically, the Atlantic Meridional Overturning Circulation: AMOC) , changing rainfall patterns in Europe and Africa, as well as affecting the Amazon, East Asia, the Southern Ocean, and ultimately accelerating Antarctic ice loss. They also define an equation to calculate the magnitude of the emergency we are facing: E = R × U = p × D × τ / T, which is an interesting way to quantify how great the emergency we face is. (Summary by Mary Williams) Nature 10.1038/d41586-019-03595-0
Biosynthesis of the nitrogenase active-site cofactor precursor NifB-co in Saccharomyces cerevisiae
 Nitrogenases are large, multisubunit enzymes that require complex metal cofactors. They are the only enzymes capable of fixing N2 into usable form, and are only produced by some archaea and bacteria; “nitrogen-fixing” legumes actually depend on the presence of their bacterial symbionts for this function. Because applying nitrogenous fertilizers to crops is environmentally costly, many efforts are underway to develop nitrogen-fixing plants. One approach would be for the plant to plant produce nitrogenase itself, but this is proving to be a difficult challenge. Previous studies have suggested that the bacteria-derived mitochondria would be the optimal place for nitrogenase production, and many of the genes encoding nitrogenase subunits have been successfully expressed in yeast mitochondria, but the resulting complex lacked the essential metal cofactor. After an impressive amount of work and starting with 25 genes of bacterial and archaeal origin, Burén et al. have now succeeded in getting this cofactor synthesized in yeast mitochondria, paving the way for formation of the complete holoenzyme. A couple of key findings were that gene sequences did not predict successful protein expression, solubility and activity, and that the successful strategy involved expressing genes from the extreme thermophile Methanothermobacter thermautotrophicus, which might account for the proteins’ greater stability in a heterologous system. (Summary by Mary Williams) Proc. Natl. Acad. Sci. USA 10.1073/pnas.1904903116
Nitrogenases are large, multisubunit enzymes that require complex metal cofactors. They are the only enzymes capable of fixing N2 into usable form, and are only produced by some archaea and bacteria; “nitrogen-fixing” legumes actually depend on the presence of their bacterial symbionts for this function. Because applying nitrogenous fertilizers to crops is environmentally costly, many efforts are underway to develop nitrogen-fixing plants. One approach would be for the plant to plant produce nitrogenase itself, but this is proving to be a difficult challenge. Previous studies have suggested that the bacteria-derived mitochondria would be the optimal place for nitrogenase production, and many of the genes encoding nitrogenase subunits have been successfully expressed in yeast mitochondria, but the resulting complex lacked the essential metal cofactor. After an impressive amount of work and starting with 25 genes of bacterial and archaeal origin, Burén et al. have now succeeded in getting this cofactor synthesized in yeast mitochondria, paving the way for formation of the complete holoenzyme. A couple of key findings were that gene sequences did not predict successful protein expression, solubility and activity, and that the successful strategy involved expressing genes from the extreme thermophile Methanothermobacter thermautotrophicus, which might account for the proteins’ greater stability in a heterologous system. (Summary by Mary Williams) Proc. Natl. Acad. Sci. USA 10.1073/pnas.1904903116
Widespread long-range cis-regulatory elements in the maize genome ($)
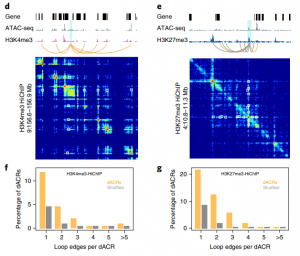 The chromatin landscape is key in the regulation of gene expression. Many studies have been made in Arabidopsis, but its genomic architecture is different to other plants. Here Ricci et al. present a very comprehensive study of chromatin analysis in maize. This work includes a huge amount of sequencing data, including ATAC-seq (transposase-accessible chromatin sequencing) chromatin accessibility landscape, DAP-seq for 32 maize transcription factor genome-wide binding sites, 9 ChiP-seq for histone features, and loop analysis by HiC and HiChiP. In maize, one-third of the accessible chromatin regions are located far from gene transcription starting sites (>2kb). These regions are enriched in transcription factor binding sites. Moreover, different correlation analysis suggests they are functional and important for gene regulation. In some cases, these distal regions form chromatin loops mediated by transcription factors and histone modifications. Putative enhancer activity assessed in protoplasts also supports the hypothesis that they are functional. Together with a companion paper by Lu et al. (see below), these works are opening doors to study distal enhancers in plants and for comparison to other eukaryotes. (Summary by Facundo Romani) Nature Plants 10.1038/s41477-019-0547-0
The chromatin landscape is key in the regulation of gene expression. Many studies have been made in Arabidopsis, but its genomic architecture is different to other plants. Here Ricci et al. present a very comprehensive study of chromatin analysis in maize. This work includes a huge amount of sequencing data, including ATAC-seq (transposase-accessible chromatin sequencing) chromatin accessibility landscape, DAP-seq for 32 maize transcription factor genome-wide binding sites, 9 ChiP-seq for histone features, and loop analysis by HiC and HiChiP. In maize, one-third of the accessible chromatin regions are located far from gene transcription starting sites (>2kb). These regions are enriched in transcription factor binding sites. Moreover, different correlation analysis suggests they are functional and important for gene regulation. In some cases, these distal regions form chromatin loops mediated by transcription factors and histone modifications. Putative enhancer activity assessed in protoplasts also supports the hypothesis that they are functional. Together with a companion paper by Lu et al. (see below), these works are opening doors to study distal enhancers in plants and for comparison to other eukaryotes. (Summary by Facundo Romani) Nature Plants 10.1038/s41477-019-0547-0
The prevalence, evolution and chromatin signatures of plant regulatory elements ($)
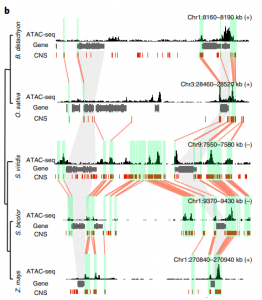 With a wide range of genome sizes and varying architectures, understanding the evolution and function of non-coding regions is not straightforward in plants. Nevertheless, many advances are coming out that will shed light on this complex scientific problem. Lu et al. performed ATAC-seq to study chromatin accessibility in 13 plant species spanning most angiosperm taxonomic groups. They found that accessible chromatin region (ACR) distribution correlates with genome size and their localization relative to transcription starting sites is very different among species. In many cases, ACRs are conserved across species, in terms of sequence and relative position to genes, even for distal ACRs (>2kb). Interestingly, transposable elements played a major role in the evolution of ACR distribution among plant genomes. They also studied chromatin features in all analyzed species and made them available online for researchers (http://epigenome.genetics.uga.edu/PlantEpigenome/). Accessible regions also possess characteristic histone modifications similarly to animal genomes but lacking H3K4me1. These chromatin states in ACRs are also conserved in different species suggesting they could be important for gene regulation. Future works will help to validate their function in planta. (Summary by Facundo Romani) Nature Plants 10.1038/s41477-019-0548-z
With a wide range of genome sizes and varying architectures, understanding the evolution and function of non-coding regions is not straightforward in plants. Nevertheless, many advances are coming out that will shed light on this complex scientific problem. Lu et al. performed ATAC-seq to study chromatin accessibility in 13 plant species spanning most angiosperm taxonomic groups. They found that accessible chromatin region (ACR) distribution correlates with genome size and their localization relative to transcription starting sites is very different among species. In many cases, ACRs are conserved across species, in terms of sequence and relative position to genes, even for distal ACRs (>2kb). Interestingly, transposable elements played a major role in the evolution of ACR distribution among plant genomes. They also studied chromatin features in all analyzed species and made them available online for researchers (http://epigenome.genetics.uga.edu/PlantEpigenome/). Accessible regions also possess characteristic histone modifications similarly to animal genomes but lacking H3K4me1. These chromatin states in ACRs are also conserved in different species suggesting they could be important for gene regulation. Future works will help to validate their function in planta. (Summary by Facundo Romani) Nature Plants 10.1038/s41477-019-0548-z
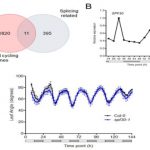
Global transcriptome analysis reveals circadian control of splicing events in Arabidopsis thaliana
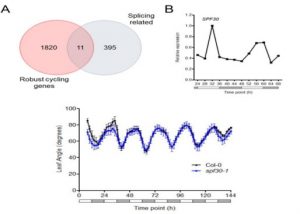 Developmental programming, metabolism and plant growth are regulated by the circadian rhythm that integrates environmental signals and plant growth. In this paper, Romanowski et al. have used the RNAseq approach to determine post-transcriptional regulation in Arabidopsis thaliana, specifically alternate splicing regulated by circadian rhythm. Tissues from 15-day old plants with altered light conditions (affecting growth), were harvested for deep sequencing. Using an in-house bioinformatics pipeline, the alternative splicing events were determined. In addition to the identification of novel regulated genes, the authors identified multiple transcripts undergoing post-transcriptional regulation. The authors also identified that the circadian rhythm regulates the spliceosome machinery in Arabidopsis by affecting the genes including SPLICING FACTOR 30 (AtSPF30). (Summary by Suresh Damodaran) bioRxiv 10.1101/845560.
Developmental programming, metabolism and plant growth are regulated by the circadian rhythm that integrates environmental signals and plant growth. In this paper, Romanowski et al. have used the RNAseq approach to determine post-transcriptional regulation in Arabidopsis thaliana, specifically alternate splicing regulated by circadian rhythm. Tissues from 15-day old plants with altered light conditions (affecting growth), were harvested for deep sequencing. Using an in-house bioinformatics pipeline, the alternative splicing events were determined. In addition to the identification of novel regulated genes, the authors identified multiple transcripts undergoing post-transcriptional regulation. The authors also identified that the circadian rhythm regulates the spliceosome machinery in Arabidopsis by affecting the genes including SPLICING FACTOR 30 (AtSPF30). (Summary by Suresh Damodaran) bioRxiv 10.1101/845560.
Dynamic ubiquitination determines transcriptional activity of the plant immune coactivator NPR1
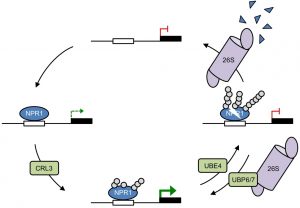 Plants (and animals) need to strike a delicate balance when activating their immune responses: not too much and not too little. The transcriptional coactivator NPR1 [nonexpressor of pathogenesis-related (PR) genes 1] has long been known to have a key role in this balance, and it itself has a complex regulation that includes regulated nuclear import and high rates of protein turnover. Now, Skelly et al. have found that this ubiquitin-mediated proteolysis also involves a delicate balance. The authors found that addition of a small number of ubiquitin monomers by the CRL3 E3 ligase activates NPR1, whereas further ubiquitination by an E4 ligase leads to its proteolysis. The authors propose “the sequential actions of E3 and E4 ligases may generate a transcriptional timer that controls the activity and lifetime of unstable (co)activators.” (Summary by Mary Williams) eLIFE 10.7554/eLife.47005
Plants (and animals) need to strike a delicate balance when activating their immune responses: not too much and not too little. The transcriptional coactivator NPR1 [nonexpressor of pathogenesis-related (PR) genes 1] has long been known to have a key role in this balance, and it itself has a complex regulation that includes regulated nuclear import and high rates of protein turnover. Now, Skelly et al. have found that this ubiquitin-mediated proteolysis also involves a delicate balance. The authors found that addition of a small number of ubiquitin monomers by the CRL3 E3 ligase activates NPR1, whereas further ubiquitination by an E4 ligase leads to its proteolysis. The authors propose “the sequential actions of E3 and E4 ligases may generate a transcriptional timer that controls the activity and lifetime of unstable (co)activators.” (Summary by Mary Williams) eLIFE 10.7554/eLife.47005
The use of high throughput phenotyping for assessment of heat stress-induced changes in Arabidopsis
 With global temperatures rising, tolerance to heat is becoming increasingly important as a breeding target for crop plants, but it is a highly complex response that includes processes including plant cooling capacity, growth recovery, and maintenance of photosynthesis. Using Arabidopsis, Gao et al. developed a pipeline to look for scorable early phenotypic responses to heat, to facilitate breeding efforts. They used a high throughput imaging system to track plant size, morphology, temperature and chlorophyll fluorescence (to calculate quantum yield) to analyze the high-temperature responses of wild-type Arabidopsis and an hsp101 non-thermotolerant mutant. They found that “early changes in photochemical quenching corresponded with the rosette size at later stages.” (Summary by Mary Williams) bioRxiv 10.1101/838102
With global temperatures rising, tolerance to heat is becoming increasingly important as a breeding target for crop plants, but it is a highly complex response that includes processes including plant cooling capacity, growth recovery, and maintenance of photosynthesis. Using Arabidopsis, Gao et al. developed a pipeline to look for scorable early phenotypic responses to heat, to facilitate breeding efforts. They used a high throughput imaging system to track plant size, morphology, temperature and chlorophyll fluorescence (to calculate quantum yield) to analyze the high-temperature responses of wild-type Arabidopsis and an hsp101 non-thermotolerant mutant. They found that “early changes in photochemical quenching corresponded with the rosette size at later stages.” (Summary by Mary Williams) bioRxiv 10.1101/838102
Evolution of carnivorous traps from planar leaves through simple shifts in gene expression
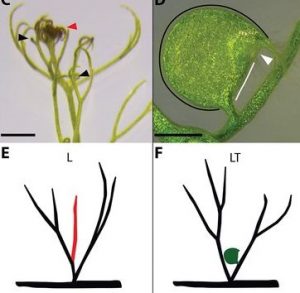 When is a leaf not a leaf? When it’s a trap. Just about everyone, including Charles Darwin, has been fascinated by carnivory in plants, which involves the development of structures that capture or trap food. Whitewoods, Gonçalves, Cheng et al. investigated how traps form in the humped bladderwort Utricularia gibba. They imaged developing leaflet and trap primordia, examined expression of genes known to control leaf shape, overexpressed one of these (UgPHV1), and based on these findings developed models the recreate that development of the traps. They found that multiple leaf forms can be generated simply by small changes in domains of gene expression, showing “how cup-shaped forms evolved multiple times independently from species with planar leaves.” (Summary by Mary Williams) Science 10.1126/science.aay5433
When is a leaf not a leaf? When it’s a trap. Just about everyone, including Charles Darwin, has been fascinated by carnivory in plants, which involves the development of structures that capture or trap food. Whitewoods, Gonçalves, Cheng et al. investigated how traps form in the humped bladderwort Utricularia gibba. They imaged developing leaflet and trap primordia, examined expression of genes known to control leaf shape, overexpressed one of these (UgPHV1), and based on these findings developed models the recreate that development of the traps. They found that multiple leaf forms can be generated simply by small changes in domains of gene expression, showing “how cup-shaped forms evolved multiple times independently from species with planar leaves.” (Summary by Mary Williams) Science 10.1126/science.aay5433
Genetic contribution of paleopolyploidy to adaptive evolution in angiosperms
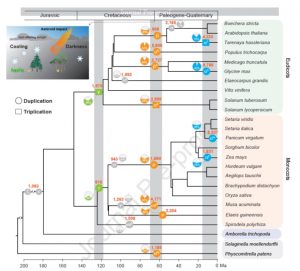 Comparative genomics has revealed that the angiosperms have experienced numerous whole-genome duplications (WGD), which have been proposed to have contributed to their global dominance. Following WGD, many of the extra gene copies are lost, but many are retained. Wu et al. explore the consequences of WGD by evaluating the gene families that have expanded following three occurrences of WGD in 25 plant genomes. They found that stress-related transcription factor families have been highly retained following WGDs, suggesting that these events have supported stress-resiliency. They also found that at the time of the Cretaceous–Paleocene (K-Pg) boundary (66 million years ago, when dinosaurs went extinct following an asteroid impact), genes involved in cold tolerance and dark tolerance were selectively retained, which is consistent with the global cooling and darkness that occurred at this time. So, the next time you’re chatting with a dinosaur-mad child, you’ve got an interesting and relevant plant story to share with them about what happened to plants after the asteroid hit. (Summary by Mary Williams) Mol. Plant 10.1016/j.molp.2019.10.012
Comparative genomics has revealed that the angiosperms have experienced numerous whole-genome duplications (WGD), which have been proposed to have contributed to their global dominance. Following WGD, many of the extra gene copies are lost, but many are retained. Wu et al. explore the consequences of WGD by evaluating the gene families that have expanded following three occurrences of WGD in 25 plant genomes. They found that stress-related transcription factor families have been highly retained following WGDs, suggesting that these events have supported stress-resiliency. They also found that at the time of the Cretaceous–Paleocene (K-Pg) boundary (66 million years ago, when dinosaurs went extinct following an asteroid impact), genes involved in cold tolerance and dark tolerance were selectively retained, which is consistent with the global cooling and darkness that occurred at this time. So, the next time you’re chatting with a dinosaur-mad child, you’ve got an interesting and relevant plant story to share with them about what happened to plants after the asteroid hit. (Summary by Mary Williams) Mol. Plant 10.1016/j.molp.2019.10.012
Shared genetic control of root system architecture between Zea mays and Sorghum bicolor
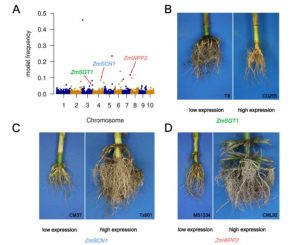 Root research got a boost from Arabidopsis grown on Petri plants, but what do you do if you study something bigger, or want to see how roots grow in soil? Zheng et al. developed a new set of tools they call CREAMD-COFE [Core Root Excavation using Compressed-air (to extract the roots from soil), and Core Root Feature Extraction (to analyse the root system)]; they argue that the use of compressed air is faster than washing roots with water, and doesn’t require a water source. They used these tools to evaluate root systems from hundreds of genotypes of Zea mays and Sorghum bicolor for SNP-based and expression-based GWAS analysis. Some of the identified genes (16%) are known to affect root system architecture in other species. They also identified seven pairs of syntenic genes from maize and sorghum that affect root system architecture. As well as demonstrating the usefulness of CREAMD-COFE, this study also shows the value of comparing GWAS data between species. (Summary by Mary Williams) Plant Physiol. 10.1104/pp.19.00752
Root research got a boost from Arabidopsis grown on Petri plants, but what do you do if you study something bigger, or want to see how roots grow in soil? Zheng et al. developed a new set of tools they call CREAMD-COFE [Core Root Excavation using Compressed-air (to extract the roots from soil), and Core Root Feature Extraction (to analyse the root system)]; they argue that the use of compressed air is faster than washing roots with water, and doesn’t require a water source. They used these tools to evaluate root systems from hundreds of genotypes of Zea mays and Sorghum bicolor for SNP-based and expression-based GWAS analysis. Some of the identified genes (16%) are known to affect root system architecture in other species. They also identified seven pairs of syntenic genes from maize and sorghum that affect root system architecture. As well as demonstrating the usefulness of CREAMD-COFE, this study also shows the value of comparing GWAS data between species. (Summary by Mary Williams) Plant Physiol. 10.1104/pp.19.00752