Fine Tuning Floral Morphology: MADS-Box Protein Complex Formation in Maize
Plant floral organ development is orchestrated by master regulatory genes that control the differentiation of meristem tissue. According to the ABC(DE) model of flower development (Bowman et al., 1989; nicely summarized by Irish, 2017), different floral organs form in concentric whorls (verticils) in response to the expression of different classes of homeotic genes. As per this model, A- and E-class genes specify sepals in the first whorl, A-, B-, and E-class genes specify petal identity in the second whorl, B-, C-, and E-class genes specify stamen identify in the third whorl, and in the fourth whorl carpel identity is specified by C- and E-class genes. All except one of the ABCDE gene classes specify MADS-box transcription factors (TFs). According to the “floral quartet” or “combinatorial assembly” model (Theissen et al. 2016), different MADS-box proteins interact with one another to form tetrameric protein complexes, with the specific composition determining which DNA sequences they bind with and thus how they regulate gene expression. For instance, stamen identity, specified by B-,C- and E-class MADS-box TFs, requires the formation of B/C/E tetramers containing B-, C-, and E-class subunits (Theissen et al 2016). This model explains the fine control over flower shape and development via the development of novel tetramer composition. However, despite its intuitive appeal, and evidence from protein-protein interaction studies that agree with its predictions, the implications of the floral quartet model have not been extensively tested in planta.
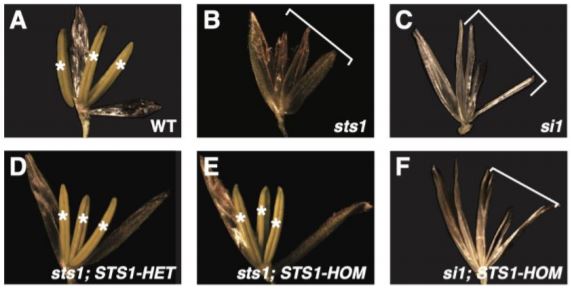
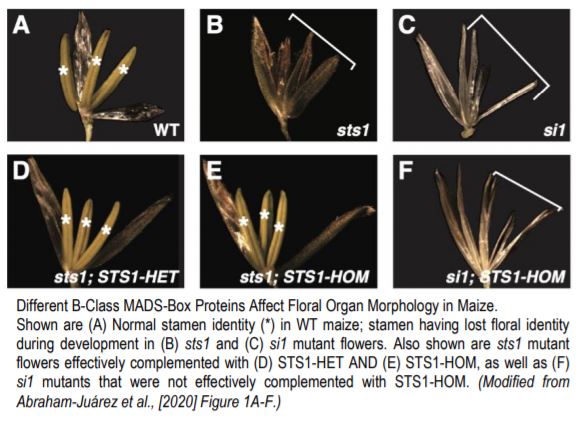 A recent paper by Abraham-Juárez et al. (2020) reports experiments using the maize B-class MADS-box protein STERILE TASSEL SILKY EAR 1 (STS1) to evaluate two key predictions of the floral quartet model: first, that components of MADS-box complexes are deeply conserved, and second, that differences in MADS-box protein-protein interactions will cause differences in gene expression that affect floral development. Normal STS1 obligately forms heterodimers with SI1 (SILKY 1), and the resulting protein plays an important role in downstream gene regulation. Here, the authors generated transgenic maize with a single amino acid change—i.e. position 81 changed from glycine to aspartic acid—that re-created an ancestral version of STS1 with a novel function, and restored its ability to form homodimers with other copies of itself. Lines carrying the normal and homodimer-forming forms of STS1 were termed STS1-HET and STS1-HOM, respectively.
A recent paper by Abraham-Juárez et al. (2020) reports experiments using the maize B-class MADS-box protein STERILE TASSEL SILKY EAR 1 (STS1) to evaluate two key predictions of the floral quartet model: first, that components of MADS-box complexes are deeply conserved, and second, that differences in MADS-box protein-protein interactions will cause differences in gene expression that affect floral development. Normal STS1 obligately forms heterodimers with SI1 (SILKY 1), and the resulting protein plays an important role in downstream gene regulation. Here, the authors generated transgenic maize with a single amino acid change—i.e. position 81 changed from glycine to aspartic acid—that re-created an ancestral version of STS1 with a novel function, and restored its ability to form homodimers with other copies of itself. Lines carrying the normal and homodimer-forming forms of STS1 were termed STS1-HET and STS1-HOM, respectively.
The authors then tested whether crossing with STS1-HET and STS1-HOM lines was able to complement A619 maize lines with null alleles for SI1 or STS1. These mutant lines—si1 and sts1—both showed no stamen or petal identity formation. However, both STS1-HET and STS1-HOM lines effectively complemented sts1, while STS1-HOM was not able to complement si1 lines. This is a crucial finding—the functional homodimer created by STS1-HOM lines cannot compensate for lost SI1 function, indicating that proper dimerization regulates MADS-box protein complex specificity. Furthermore, the flowers of STS1-HOM lines showed wider and shorter anthers than STS1-HET flowers. The authors also observed substantial differences in protein (but not RNA) localization, with STS1-HOM, but not STS1-HET, localizing to gynoecia. Transcriptomic analyses found that STS1-HOM plants showed a higher number of differentially expressed genes (DEGs) than STS1-HET plants, but also that 29 of 65 GO terms regulated by both STS1-HOM and STS1-HET were related to development. However, immunoprecipitation-mass spectroscopy (IP-MS) analysis of proteins bound to STS1-HOM and STS1-HET revealed that less than 10% of the sets of proteins identified as interactors of either were shared, thereby confirming their distinct roles. Further analysis of IP-MS data revealed that MADS-box proteins were four to seven times more abundant in STS1-HOM plants than in STS1-HET plants, indicating that STS1-HOM protein was more likely to accumulate in floral tissue, despite comparable levels of gene expression. This suggested post-translational modification of MADS-box proteins related to STS1 homodimerization. Both proteins co-precipitated with proteins related to ubiquitination. However, no clear differences in ubiquitination or phosphorylation were identified between STS1-HOM and STS-HET proteins, and therefore post-translational differences resulting in differences in protein degradation may be subtle and/or indirect.
In conclusion, these data provide new evidence that differential B-class protein dimerization can result in quantitative differences in floral morphology. This is significant because continuous or subtle differences in organ shape are not as well understood as categorical changes in floral organ identity. The authors also provide strong evidence that the functions of B-, C-, and E-class protein complexes are deeply conserved, even between monocots and dicots. Finally, although the precise mechanism by which post-translational differences remains obscure, the findings reported here provide a basis on which to conduct future studies.
William Hughes
Department of Ecology, Environment, and Plant Sciences,
Stockholm University
Svante Arrhenius väg 20
114 18 Stockholm
ORCID: 0000-0003-4142-5279
REFERENCES
Abraham-Juárez, M., Schrager-Lavelle, A.,Man, J., Whipple, C., Handakumbura, P.,Babbitt, C., and Bartlett, M. 2020. Evolutionary variation in MADS-box dimerization affects floral development and protein abundance in maize. Plant Cell. Sept 2020. DOI: 10.1105/tpc.20.00300
Bowman, J., Smyth, D., Meyerowitz, E. 1989. Genes directing flower development in Arabidopsis. Plant Cell. 1(1): 37-52; DOI: 10.1105/tpc.1.1.37
Irish, V. 2017. The ABC model of floral development. Current Biology 27, R853-R909. DOI: 10.1016/j.cub.2017.03.045
Theissen, G., Melzer, R., and Rümpler, F. (2016). MADS-domain transcription factors and the floral quartet model of flower development: linking plant development and evolution. Devel. 143: 3259–3271. DOI: 10.1242/dev.134080