Chlamydomonas keeps the rhythm going
By Patrice Salomé, Department of Chemistry and Biochemistry, UCLA, Los Angeles, CA
Background: Transcriptome deep sequencing (RNA-seq) has become a routine method to query changes in gene expression after a genetic, physiological or chemical perturbation. While each laboratory typically analyzes a few samples in their condition of choice, the community has explored the transcriptional landscape of countless species under hundreds, if not thousands, of variables.
Question: We wished to summarize the past ten years of RNA-seq data from the unicellular green alga Chlamydomonas reinhardtii into a single, large, normalized expression matrix for co-expression analysis. We reasoned that the various growth conditions and treatments queried by each individual study would modulate the expression of all genes in a genetically programmed manner, which would become accessible by looking for co-expressed genes with a gene or genes of interest.
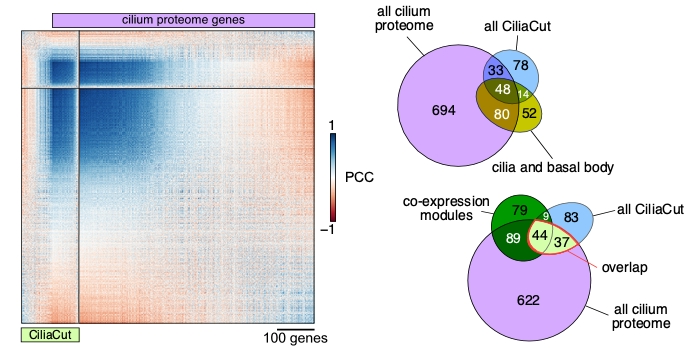
Findings: Depending on the level of stringency applied, any given nuclear gene in Chlamydomonas reinhardtii is co-expressed with tens to hundreds of genes. We determined that about 400 genes (out of a 1,000 predicted to encode cilia components based on a published proteomics data set) were strongly co-expressed. In addition, these co-expressed genes shared the same diurnal phase, matching the time when cells regenerated their flagella after cell division. We applied the same analysis to genes with roles in cell division, photosynthesis, tetrapyrrole biosynthesis and respiration, yielding hundreds of promising candidate genes for follow-ups. Unexpectedly, we also observed a strong rhythmic component (diurnal, circadian, or driven by the cell cycle) in most RNA-seq samples, although the sequenced RNA was extracted from cells grown in constant light. These results indicate that cells may remain synchronized for far longer than previously assumed, stressing the need to collect time-matched samples for RNA-seq rather than waiting for cultures to reach equal cell densities.
Next steps: Our approach rapidly generates a list of high-confidence candidate genes based on user-defined criteria. Such lists can then be compared to existing data obtained from other methods such as proteomics or large-scale mutant screens being carried out in Chlamydomonas reinhardtii. The overlap between each list may not be perfect, but might make us feel more confident in our choice of genes for follow-up studies.
Reference:
Patrice A. Salomé and Sabeeha S. Merchant (2021). Co-Expression Networks in Chlamydomonas Reveal Significant Rhythmicity in Batch Cultures and Empower Gene Function Discovery. Plant Cell, https://doi.org/10.1093/plcell/koab042







