What We’re Reading: October 19th
 This week’s edition is guest edited by Matthias Benoit, a postdoctoral Research Associate at The Sainsbury Laboratory University of Cambridge. His research focuses on the developmental, environmental and epigenetic regulation of tomato retrotransposons. His favorite models of study are fruit development and ripening, and abiotic stresses. He’s also engaged in teaching and communicating plant sciences. Plantae Profile.
This week’s edition is guest edited by Matthias Benoit, a postdoctoral Research Associate at The Sainsbury Laboratory University of Cambridge. His research focuses on the developmental, environmental and epigenetic regulation of tomato retrotransposons. His favorite models of study are fruit development and ripening, and abiotic stresses. He’s also engaged in teaching and communicating plant sciences. Plantae Profile.
Review: The role of plant epigenetics in biotic interactions
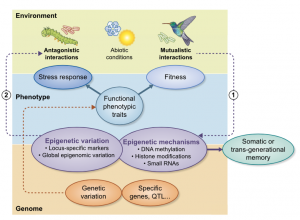 Plant phenotypes are influenced by the nature and intensity of biotic interactions. While the role of genetic diversity has been extensively studied, contribution of epigenetics to plant fitness and response to biotic stresses remains elusive. The authors review here the most recent literature about the contribution of epigenetic marks such as DNA methylation and histone post-translational marks to the regulation of plant biotic interactions. They stress-out the bi-directionality of these interactions, discussing experimental evidences about (1) how biotic interactors modulate the plant’s epigenetic landscape and (2) how the plant’s epigenetic configuration influences the response to the biotic stress. The authors highlight the need for a better integration of ecology, molecular biology and genomics in order to harness biotic interaction-induced epigenetic variation for phenotypic traits improvement. (Summary by Matthias Benoit).
Plant phenotypes are influenced by the nature and intensity of biotic interactions. While the role of genetic diversity has been extensively studied, contribution of epigenetics to plant fitness and response to biotic stresses remains elusive. The authors review here the most recent literature about the contribution of epigenetic marks such as DNA methylation and histone post-translational marks to the regulation of plant biotic interactions. They stress-out the bi-directionality of these interactions, discussing experimental evidences about (1) how biotic interactors modulate the plant’s epigenetic landscape and (2) how the plant’s epigenetic configuration influences the response to the biotic stress. The authors highlight the need for a better integration of ecology, molecular biology and genomics in order to harness biotic interaction-induced epigenetic variation for phenotypic traits improvement. (Summary by Matthias Benoit).
New Phytologist 10.1111/nph.15408
Review: Spontaneous epimutations in plants
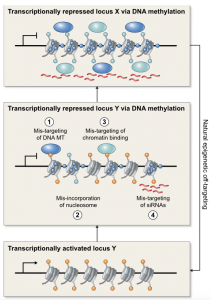 Spontaneous epimutations are stochastic and heritable changes of DNA methylation in genomes. In this review, Johannes and Schmitz discuss the molecular origins of spontaneous epimutations in plants as well as their functional and phenotypical consequences. The authors highlight that spontaneous epimutations are frequent in plant genomes and are likely induced by events interfering with the activity of enzymes involved in establishing or maintaining DNA methylation. Evidences for heritable phenotypic changes induced by spontaneous epimutations remain limited but are linked to changes in expression of the surrounding genes. Further characterization of the epimutational processes is the next challenge to understand in which extent epimutations shaped plant phenotypes over evolutionary times. (Summary by Matthias Benoit).
Spontaneous epimutations are stochastic and heritable changes of DNA methylation in genomes. In this review, Johannes and Schmitz discuss the molecular origins of spontaneous epimutations in plants as well as their functional and phenotypical consequences. The authors highlight that spontaneous epimutations are frequent in plant genomes and are likely induced by events interfering with the activity of enzymes involved in establishing or maintaining DNA methylation. Evidences for heritable phenotypic changes induced by spontaneous epimutations remain limited but are linked to changes in expression of the surrounding genes. Further characterization of the epimutational processes is the next challenge to understand in which extent epimutations shaped plant phenotypes over evolutionary times. (Summary by Matthias Benoit).
New Phytologist 10.1111/nph.15434
Non-CG methylation mechanism in Marchantia polymorpha ($)
 Previous reports have shown that M. polymorpha DNA methylation status varies during its life cycle and it contains only one copy of the METHYLTRANSFERASE1 (MET1) orthologous gene. MET1 is responsible of CG methylation in angiosperms. Ikeda and collaborators generated MpMET mutants using the CRISPR/CAS9 system. The mutants showed abnormal shape, growth delay, reduced size and organs for asexual and sexual reproduction were not formed. Further analysis showed DNA hypomethylation of transposon regions. Bisulfite sequencing (BS-seq) was used to analyze wide genome methylation in the mutants; finding a 0,5% decreased methylation compared to wild type. Non-CG methylation was increased in transposon elements (TE) in mutants. Furthermore, CAG methylation was more efficient compared to CTG in the wild type, resulting on a asymmetrical methylation. On the other hand, the lack of CG methylation in the mutants resulted in a symmetrical methylation of CAG and CTG sites. (Summary by Cecilia Vasquez-Robinet).
Previous reports have shown that M. polymorpha DNA methylation status varies during its life cycle and it contains only one copy of the METHYLTRANSFERASE1 (MET1) orthologous gene. MET1 is responsible of CG methylation in angiosperms. Ikeda and collaborators generated MpMET mutants using the CRISPR/CAS9 system. The mutants showed abnormal shape, growth delay, reduced size and organs for asexual and sexual reproduction were not formed. Further analysis showed DNA hypomethylation of transposon regions. Bisulfite sequencing (BS-seq) was used to analyze wide genome methylation in the mutants; finding a 0,5% decreased methylation compared to wild type. Non-CG methylation was increased in transposon elements (TE) in mutants. Furthermore, CAG methylation was more efficient compared to CTG in the wild type, resulting on a asymmetrical methylation. On the other hand, the lack of CG methylation in the mutants resulted in a symmetrical methylation of CAG and CTG sites. (Summary by Cecilia Vasquez-Robinet).
Plant & Cell Physiology 10.1093/pcp/pcy161
Stage-specific transcriptomes and DNA methylomes indicate an early and transient loss of transposon control in Arabidopsis shoot stem cells
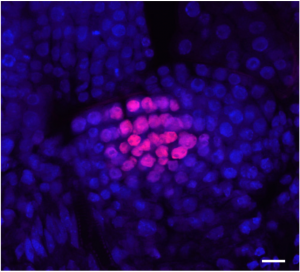 Postembryonic development in plants relies on stem cells located within the shoot apical meristem. Male and female gametes are descendants of those stem cells and maintenance of genome stability in this pool of cells is thus fundamental. In-depth molecular analysis has so far been hindered by the tedious isolation of shoot apical meristem stem cells. Gutzat et al. generated a development stage series of transcriptomes from stem cells isolated using fluorescence nuclear sorting. Together with stem-cell specific genes, the authors report the increased expression of transposons in stem cells. This is correlated with changes in DNA methylation at transposons occurring prior the commitment of the stem cells to the reproductive lineage. This suggests an early event of epigenetic reprogramming, and opens new perspectives for the study of epigenetic heritability. (Summary by Matthias Benoit).
Postembryonic development in plants relies on stem cells located within the shoot apical meristem. Male and female gametes are descendants of those stem cells and maintenance of genome stability in this pool of cells is thus fundamental. In-depth molecular analysis has so far been hindered by the tedious isolation of shoot apical meristem stem cells. Gutzat et al. generated a development stage series of transcriptomes from stem cells isolated using fluorescence nuclear sorting. Together with stem-cell specific genes, the authors report the increased expression of transposons in stem cells. This is correlated with changes in DNA methylation at transposons occurring prior the commitment of the stem cells to the reproductive lineage. This suggests an early event of epigenetic reprogramming, and opens new perspectives for the study of epigenetic heritability. (Summary by Matthias Benoit).
bioRxiv 2018/10/05/430447
DNA methylation footprints during soybean domestication and improvement
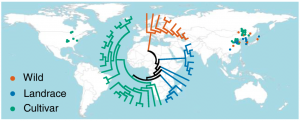 Crop domestication relies on harnessing the natural genomic diversity present in the cultivated population. The contribution of genetic variation for selection and improvement of traits in plants has been extensively studied. Besides this, variation in epigenetic components such as DNA methylation offers an alternative and mostly independent source of variability, but the contribution of epigenetic variation to crop domestication remains elusive. To assess this, the authors provided here methylomes for 45 soybean accessions, including wild soybeans, landraces and cultivars. They identified more than 5000 differentially methylated regions, mostly independent of loci showing genetic variation. Intriguingly, a significant fraction of differentially methylated regions are associated with genes involved in the carbohydrate pathways. This reveals the importance of epigenetic variation in soybean domestication and trait improvement. (Summary by Matthias Benoit).
Crop domestication relies on harnessing the natural genomic diversity present in the cultivated population. The contribution of genetic variation for selection and improvement of traits in plants has been extensively studied. Besides this, variation in epigenetic components such as DNA methylation offers an alternative and mostly independent source of variability, but the contribution of epigenetic variation to crop domestication remains elusive. To assess this, the authors provided here methylomes for 45 soybean accessions, including wild soybeans, landraces and cultivars. They identified more than 5000 differentially methylated regions, mostly independent of loci showing genetic variation. Intriguingly, a significant fraction of differentially methylated regions are associated with genes involved in the carbohydrate pathways. This reveals the importance of epigenetic variation in soybean domestication and trait improvement. (Summary by Matthias Benoit).
Genome Biology 10.1186/s13059-018-1516-z
Insertion of a transposon-like sequence in the 5’-flanking region of the YUCCA gene causes the stony hard phenotype ($)
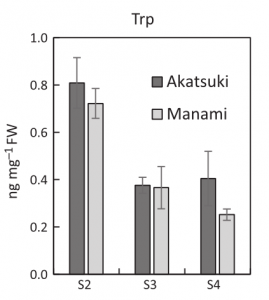 Fruit softening in melting-flesh peaches is triggered by a major accumulation of ethylene at the late stage of ripening. Existence of stony hard peaches showing inhibition of fruit softening has been correlated with low levels of indole-3-acetic-acid inducing low levels of ethylene, but the underlying molecular cause remains unknown. Using ultra-performance liquid chromatography and tandem mass spectrometry, the authors identified a defect in indole-3-pyruvic-acid accumulation, an indole-3-acetic-acid intermediate. This originates from the loss of YUCCA gene expression, encoding the enzyme mediating the conversion from indole-3-pyruvic-acid to indole-3-acetic-acid. The authors have identified a homozygous transposon insertion located in the YUCCA flanking region found uniquely in stony hard peaches, and observed co-segregation of the insertion with the phenotype, pinpointing YUCCA loss of expression as the main cause of the softening defect. (Summary by Matthias Benoit).
Fruit softening in melting-flesh peaches is triggered by a major accumulation of ethylene at the late stage of ripening. Existence of stony hard peaches showing inhibition of fruit softening has been correlated with low levels of indole-3-acetic-acid inducing low levels of ethylene, but the underlying molecular cause remains unknown. Using ultra-performance liquid chromatography and tandem mass spectrometry, the authors identified a defect in indole-3-pyruvic-acid accumulation, an indole-3-acetic-acid intermediate. This originates from the loss of YUCCA gene expression, encoding the enzyme mediating the conversion from indole-3-pyruvic-acid to indole-3-acetic-acid. The authors have identified a homozygous transposon insertion located in the YUCCA flanking region found uniquely in stony hard peaches, and observed co-segregation of the insertion with the phenotype, pinpointing YUCCA loss of expression as the main cause of the softening defect. (Summary by Matthias Benoit).
Plant Journal 10.1111/tpj.14070
Sequestration of a transposon-derived siRNA by a target mimic imprinted gene induces postzygotic reproductive isolation in Arabidopsis ($)
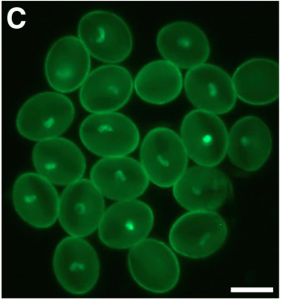 Hybridization of plants with distinct chromosome number often results in seed development defects due to a phenomenon known as the triploid block. While this is triggered by the unbalanced expression of some paternally and maternally imprinted alleles, the molecular basis of the triploid block is not fully understood. The authors describe here that the paternally expressed gene PEG2 behaves as a small RNA sponge by limiting the accumulation of the transposon-derived small interfering RNA siRNA854 in the endosperm. Upon interploidy hybridization, abnormally high sequestration of siRNA854 due to PEG2 up regulation results in aberrant accumulation of siRNA854 natural targets such as UBP1 and triggers the triploid block. This work highlights the role of transposon-derived small RNAs in reproductive isolation and speciation. (Summary by Matthias Benoit).
Hybridization of plants with distinct chromosome number often results in seed development defects due to a phenomenon known as the triploid block. While this is triggered by the unbalanced expression of some paternally and maternally imprinted alleles, the molecular basis of the triploid block is not fully understood. The authors describe here that the paternally expressed gene PEG2 behaves as a small RNA sponge by limiting the accumulation of the transposon-derived small interfering RNA siRNA854 in the endosperm. Upon interploidy hybridization, abnormally high sequestration of siRNA854 due to PEG2 up regulation results in aberrant accumulation of siRNA854 natural targets such as UBP1 and triggers the triploid block. This work highlights the role of transposon-derived small RNAs in reproductive isolation and speciation. (Summary by Matthias Benoit).
Developmental Cell S1534580718305999
DNA methylome analysis provides evidence that the expansion of the tea genome is linked to TE bursts
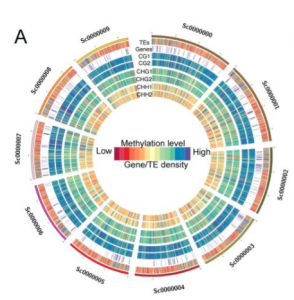 Expansion of plant genomes is thought to be linked to massive bursts of transposable elements activity. This implies consequences on the distribution of epigenetic modifications required for the silencing of the causative transposons. Consequences of transposon bursts have been extensively described in compact plant genomes such as Arabidopsis or rice, but similar analyses in very large, complex plant genomes are still lacking. Here, the authors provide the DNA methylome of the complex tea genome. They observed that the high transposon content in tea is correlated with higher levels of DNA methylation in the CHG context compared to other crops. They also show that densely methylated transposon-like sequences are found enriched in the vicinity of genes involved in metabolic pathways of agronomical interest, such as caffeine and theanine synthesis, likely influencing their expression levels. (Summary by Matthias Benoit).
Expansion of plant genomes is thought to be linked to massive bursts of transposable elements activity. This implies consequences on the distribution of epigenetic modifications required for the silencing of the causative transposons. Consequences of transposon bursts have been extensively described in compact plant genomes such as Arabidopsis or rice, but similar analyses in very large, complex plant genomes are still lacking. Here, the authors provide the DNA methylome of the complex tea genome. They observed that the high transposon content in tea is correlated with higher levels of DNA methylation in the CHG context compared to other crops. They also show that densely methylated transposon-like sequences are found enriched in the vicinity of genes involved in metabolic pathways of agronomical interest, such as caffeine and theanine synthesis, likely influencing their expression levels. (Summary by Matthias Benoit).
Plant Biotechnology Journal 10.1111/pbi.13018
Partial maintenance of organ-specific epigenetic marks during plant asexual reproduction leads to heritable phenotypic variation ($)
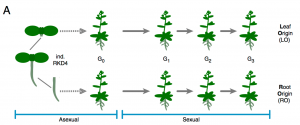 Clonal propagation is widely used by humans to maintain and propagate desirable phenotypic traits in plants. Despite a restricted genetic variety, phenotypical difference, known a somaclonal variation, arises sometimes between parents and the clonally propagated progeny. Some of this variability is attributed to epigenetic variation, but the origins and underlying molecular principles remain unclear. By artificial induction of the ectopic program from either Arabidopsis leaf or root tissue, the authors describe that tissue-specific DNA methylation is partially maintained in the whole clonal progeny and stably transmitted through meiosis. The heritability of tissue-specific DNA methylation is associated with the partial preservation of associated gene expression patterns, translating in new heritable phenotypes. This represents an original strategy to induce trait variation. (Summary by Matthias Benoit).
Clonal propagation is widely used by humans to maintain and propagate desirable phenotypic traits in plants. Despite a restricted genetic variety, phenotypical difference, known a somaclonal variation, arises sometimes between parents and the clonally propagated progeny. Some of this variability is attributed to epigenetic variation, but the origins and underlying molecular principles remain unclear. By artificial induction of the ectopic program from either Arabidopsis leaf or root tissue, the authors describe that tissue-specific DNA methylation is partially maintained in the whole clonal progeny and stably transmitted through meiosis. The heritability of tissue-specific DNA methylation is associated with the partial preservation of associated gene expression patterns, translating in new heritable phenotypes. This represents an original strategy to induce trait variation. (Summary by Matthias Benoit).







