Sequencing Plant Transcriptomes at Single-Cell Resolution Allows Unprecedented Characterization of Genetic and Developmental Cellular Processes
Jose M Celedon 1
1 Michael Smith Laboratories, The University of British Columbia, 2185 East Mall, Vancouver, BC V6T 1Z4, Canada.
Transcriptomic studies in plants usually involve anonymous survey-style methods, where whole organs or tissues are homogenized, and each cell’s contribution to transcript abundance becomes unidentifiable. This type of approach, although useful for addressing many biological questions at the organ or tissue level, is inherently limited in dissecting processes that take place in rare cell types or individual cells. Methods to profile transcripts from plant cell types were developed soon after microarrays were first used, however, they are still based on laborious and challenging techniques, including laser microdissection of morphologically distinct cell types, and tissue protoplasting followed by fluorescence-activated cell sorting of GFP-tagged cell types (Nakazono et al., 2003; Brady et al., 2007; Celedon et al., 2017). Developing single-cell RNA sequencing (scRNAseq) in plants has been markedly more challenging than in animals, due to the cell wall polymer surrounding each plant cell and the large variation in cell size.
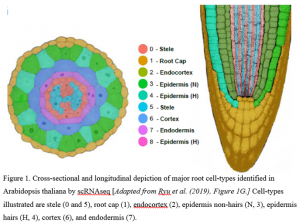 Next-generation sequencing has transformed many fields of plant biology, and recent advances in cDNA library preparation, barcoding, and protocols to isolate individual cells have made scRNAseq more feasible and less costly. In this issue of Plant Physiology, Ryu et al. (2019) describe the application of a commercially available microfluidic approach (10xGenomics) to barcode the RNA from individual cells in Arabidopsis thaliana roots and obtain >7,000 single-cell transcriptomes. Taking advantage of an Arabidopsis catalog of root cell type-specific markers, Ryu and colleagues disentangled eight major root cell type clusters (Fig. 1) and several sub-clusters, including two quiescent center cells, the rarest cell type in roots. The authors also noticed that expression levels of cell type markers varied within each cluster, with lower expression in the center and higher expression towards the edge of the cluster. This pattern suggested that cells in a cluster are organized by developmental gradients and may, in future, allow the reconstruction of plant developmental trajectories, as has been done in other organisms (Shin et al., 2015).
Next-generation sequencing has transformed many fields of plant biology, and recent advances in cDNA library preparation, barcoding, and protocols to isolate individual cells have made scRNAseq more feasible and less costly. In this issue of Plant Physiology, Ryu et al. (2019) describe the application of a commercially available microfluidic approach (10xGenomics) to barcode the RNA from individual cells in Arabidopsis thaliana roots and obtain >7,000 single-cell transcriptomes. Taking advantage of an Arabidopsis catalog of root cell type-specific markers, Ryu and colleagues disentangled eight major root cell type clusters (Fig. 1) and several sub-clusters, including two quiescent center cells, the rarest cell type in roots. The authors also noticed that expression levels of cell type markers varied within each cluster, with lower expression in the center and higher expression towards the edge of the cluster. This pattern suggested that cells in a cluster are organized by developmental gradients and may, in future, allow the reconstruction of plant developmental trajectories, as has been done in other organisms (Shin et al., 2015).
Furthermore, Ryu and co-workers showed that scRNAseq can also provide novel insight into the effect of gene mutations in cell development. As proof of concept, the authors performed scRNAseq of Arabidopsis plants with either a mutated rhd6 gene, which are unable to form root hairs, or a mutated gl2 gene, which lack non-hair cells (Masucci & Schiefelbein, 1994; Masucci et al., 1996). Analysis of root cell-type markers in these two mutant lines showed that rhd6 roots, as expected, had a significantly reduced number of hair cells and an increased number of non-hair cells, while gl2 roots showed the opposite pattern. This suggested that cells were converted from hair to non-hair and vice versa. A closer look at the transcriptomes of hair and non-hair cell clusters revealed that despite the mutated rhd6 and gl2 genes, “converted” cells showed some expression of their original cell-type markers. This implies that rhd6 and gl2 mutations do not entirely convert one cell type to the other and that other genes may contribute to defining hair and non-hair cells in Arabidopsis roots.
Transcriptome studies in model and non-model plants will greatly benefit from the increased resolution of scRNAseq. The ability to interrogate the transcriptomes of individual cells will allow a better understanding of many fundamental plant processes, including organ formation in meristems, the formation and function of the Casparian band in root endoderm cells, and the production of specialized metabolites, all of which take place in specific cell types and at specific developmental stages. Future improvements in protoplast isolation would allow the application of scRNAseq in more mature and recalcitrant plant tissues including wood, bark and seed tissues. Furthermore, increasing per-cell sequencing-depth in scRNAseq experiments may allow de novo transcriptome assemblies for each cell type, greatly facilitating gene discovery and characterization in plant species that lack sequenced genomes, but have ecological, agricultural, or industrial value.
References
Brady SM, Orlando D a, Lee J-Y, Wang JY, Koch J, Dinneny JR, Mace D, Ohler U, Benfey PN. 2007. A high-resolution root spatiotemporal map reveals dominant expression patterns. Science 318: 801–6.
Celedon JM, Yuen MMS, Chiang A, Henderson H, Reid KE, Bohlmann J. 2017. Cell-type- and tissue-specific transcriptomes of the white spruce (Picea glauca) bark unmask fine-scale spatial patterns of constitutive and induced conifer defense. Plant Journal 92: 710–726.
Masucci JD, Rerie W., Foreman D., Zhang M, Galway ME, Marks MD, Schiefelbein JW. 1996. The homeobox gene GLABRA2 is required for position-dependent cell differentiation in the root epidermis of Arabidopsis thaliana. Development 122: 1253–1260.
Masucci JD, Schiefelbein JW. 1994. The rhd6 Mutation of Arabidopsis thaliana Alters Root-Hair Initiation through an Auxin- and Ethylene-Associated Process. Plant Physiology 106: 1335–1346.
Nakazono M, Qiu F, Borsuk LA, Schnable PS. 2003. Laser-Capture Microdissection , a Tool for the Global Analysis of Gene Expression in Specific Plant Cell Types : Identification of Genes Expressed Differentially in Epidermal Cells or Vascular Tissues of Maize. Plant Cell 15: 583–596.
Shin J, Berg DA, Zhu Y, Shin JY, Song J, Bonaguidi MA, Enikolopov G, Nauen DW, Christian KM, Ming GL, et al. 2015. Single-Cell RNA-Seq with Waterfall Reveals Molecular Cascades underlying Adult Neurogenesis. Cell Stem Cell 17: 360–372.

