Another Step Closer to Understanding Plant Cell Wall Biosynthesis: The Crystal Structure of FUCOSYLTRANSFERASE1[
IN BRIEF by Nancy R. Hofmann [email protected]
Plant cell walls consist of cellulose microfibrils embedded in a matrix of polymers including hemicelluloses. As one of the main hemicelluloses in the cell walls of dicots, xyloglucan is an important target of study to understand plant cell walls in general and for polymer applications in biotechnology. Xyloglucan consists of a β-1,4-linked backbone of glucosyl residues with or without side chains, the most common of which is a xylosyl residue. The side chains themselves can harbor further side chains, and, ultimately, 24 different side chains can make up xyloglucan. Biosynthesis of xyloglucan (reviewed in Pauly and Keegstra, 2016) involves members of the glycosyltransferase (GT) superfamily, including a fucosyltransferase, which was the first plant cell wall-biosynthetic enzyme discovered. Now, in a Breakthrough Report from Rocha et al. (2016), Arabidopsis thaliana FUCOSYLTRANSFERASE1 (FUT1) provides the first crystal structure of a plant cell wall biosynthesis enzyme.
FUT1 transfers fucose from the donor GDP-fucose to galatosyl residues on xyloglucan and is a member of glycosyltransferase family 37 (GT37). Rocha and coworkers solved the crystal structures of the soluble portion of FUT1 both in the apo form and bound to GDP and a xyloglucan oligosaccharide acceptor. The most similar known structures were those of fucosyltransferases from other GT families, including Caenorhabditis elegans POFUT1 and human POFUT2, which mediate O-fucosylation of proteins in those species. These related structures have a GT-B fold, which canonically includes two α/β/α Rossmann-fold domains, termed the N- and C-domains, with the active site in a cleft between them. The FUT1 structure represents a GT-B variant not previously observed (see figure). Instead of the α/β/α N-domain, the FUT1 N-domain has a β-sheet with α-helices on one side, but loops on the other. The C-domain has a more typical Rossmann fold, but an extra C-terminal domain forms β-hairpins that lie against the N-domain. This extra domain contacts both the N- and C-domains and forms part of the acceptor binding site.
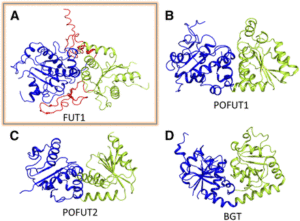
FUT1 adopts a variant of the GT-B superfamily fold. (A) FUT1 structure reported in this work. (B) and (C) Two of the most similar reported structures: (B) POFUT1 (PDB code 3ZY6) and (C) POFUT2 (PDB code 4AP5). (D) Canonical GT-B fold of the phage T4-glucosyltransferase (BGT; PDB code 1BGT), with two similar Rossmann domains: the N-domain (blue) and the C-domain (green). (Adapted from Rocha et al. [2016], Supplemental Figure 1.)
The authors also use the structure to explain FUT1’s substrate specificity. In contrast to the second galactosyl in the acceptor subunit, the first residue is on the opposite side of the β-1,4-d-glucan backbone and does not make any contact with FUT1. Accordingly, FUT1 fucosylates the second, but not the first, galactosyl residue in a xyloglucan acceptor. The extra C-terminal domain of FUT1 also interacts with acceptor. Interestingly, this domain is present in all 10 Arabidopsis thaliana GT37 family members, among which only FUT1 acts on xyloglucan. Rocha et al. were able to divide this family into three subgroups, based on their sequences in the region that contributes to the anchoring of the xyloglucan subunit in FUT1. Their analysis provides an opening into predictions of substrate specificity based on primary sequence, which has proved quite difficult among GT family members.
Overall, this work adds another first to the rich history of fucosyltransferase research in plants and provides insight into the biosynthesis of an important plant cell wall component as well as into the function of the huge family of glycosyltransferases in general.
REFERENCES
Pauly, M., and Keegstra, K. (2016). Biosynthesis of the plant cell wall matrix polysaccharide xyloglucan. Annu. Rev. Plant Biol. 67: 235–259.
Rocha, J., Cicéron, F., de Sanctis, D., Lelimousin, M., Chazalet, V., Lerouxel, O., and Breton, C. (2016). Structure of Arabidopsis thaliana FUT1 reveals a variant of the GT-B class fold and provides insight into xyloglucan fucosylation. Plant Cell 28: 2352–2364.









 Carotenoids are nutritionally important phytonutrients. Comas et al. describe a strategy to enhance the production of cartotenoids in the seed endosperm. They start with quantitative metabolomics and gene expression data which they feed into mathematical models to determine which gene(s) need to be engineered. Some of the genes they identify have been identified and verified previously, but others are newly identified as important for enhancing carotenoid biosynthesis. Plant J.
Carotenoids are nutritionally important phytonutrients. Comas et al. describe a strategy to enhance the production of cartotenoids in the seed endosperm. They start with quantitative metabolomics and gene expression data which they feed into mathematical models to determine which gene(s) need to be engineered. Some of the genes they identify have been identified and verified previously, but others are newly identified as important for enhancing carotenoid biosynthesis. Plant J. 

