Examination of Protein Complexes Gets SiMPull
IN BRIEF by [email protected]
Assessing protein-protein interactions remains a fundamental challenge for plant biologists. Current methods such as coimmunoprecipitation (co-IP), yeast two-hybrid, bimolecular fluorescence complementation (BiFC), and others can produce artifacts and also yield only a bulk “ensemble” readout that is difficult to quantify, much less examine statistically. For example, the fluorescent protein halves used in BiFC can self-assemble, and their reassociation to provide fluorescent complementation occurs irreversibly. Therefore, a BiFC signal can mislead us; BiFC requires well-chosen controls, preferably using mutated proteins and quantitation of fluorescence (for more, see Horstman et al., 2014). Also, yeast two-hybrid assays only examine pairwise interactions; if complex formation requires an intermediary, these assays might fail to detect the interaction.
Recent advances in microscopy have enabled imaging of single molecules (also termed single-particle imaging) using TIRF (total internal reflection fluorescence) microscopy; these techniques have advanced our understanding of the dynamics of membrane proteins, for example, in plants and other systems. Work in animal systems has extended these single-particle imaging methods to examine protein-protein interactions by a technique termed single-molecule pull-down (SiMPull; Jain et al., 2011). This technique uses immunoprecipitation (the Pull) to immobilize a molecule of interest (along with any interacting proteins). SiMPull then uses single-molecule imaging (the SiM) to visualize a fluorescent signal from the molecule of interest and its potential interacting partner (see workflow in figure). TIRF microscopy can image individual particles only within a narrow evanescent field and thus requires a planar field of analysis, so proteins are immobilized on a slide. Each fluorescent protein produces an individual signal and quantitating the coincidence of two signals provides statistical evidence for protein-protein interaction. Moreover, counting fluorophore photobleaching steps can reveal the stoichiometry of the complex.
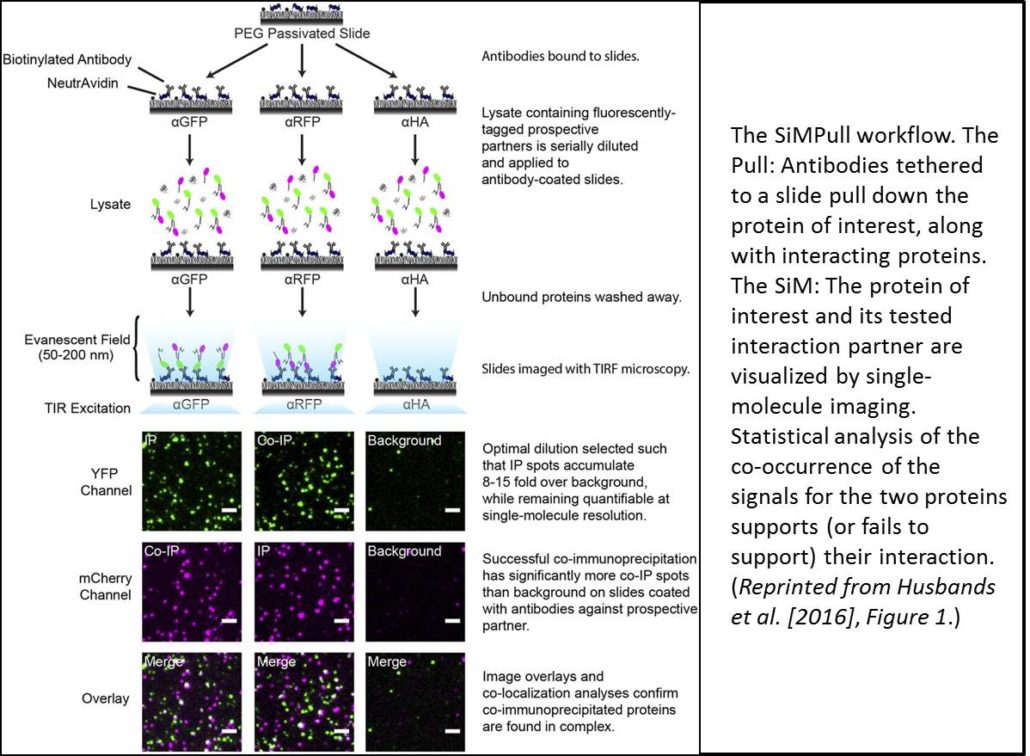
For SiMPull, proteins must be loaded onto the assay at the appropriate dilution—too little and proteins are indistinguishable from background, too much and protein signals begin to overlap. To determine the appropriate signal density, cell extracts are applied to slides as serial dilutions. However, the relative levels of each interaction partner must also be balanced; indeed, the authors found that the cells produced much more ZPR3-mCherry than PHB-YFP despite both being expressed from the 35S promoter. Examination of multiple proteins revealed that smaller proteins (such as ZPR3-mCherry) accumulate faster than larger proteins (such as PHB-YFP). To balance protein levels, the authors then expressed the larger protein under the control of a stronger promoter, the double 35S promoter (2x35S). They also examined protein accumulation at different time points and picked the time where protein levels were nearly equivalent.
Fluorophore maturation probabilities (that is, the fraction of the YFP or mCherry proteins that are actually fluorescent) can affect estimates of colocalization. If the YFP or mCherry of one partner fails to fluoresce, then the proteins will not count as colocalized. Indeed, work in mammalian systems showed maturation probabilities of ∼0.8 for YFP and 0.4 for mCherry. Therefore, the authors determined the maturation probabilities of YFP and mCherry in their system using SiMPull on tandem dimers. If both partners are fluorescent, then a YFP-YFP fusion protein should photobleach in two steps. Using the number of tandem dimers with two-step photobleaching, the authors calculated maturation probabilities of ∼0.4 for YFP and 0.55 for mCherry, values substantially different from those observed in animal cells.
Using lysates from the cells coexpressing 35S:ZPR3-mCherry and 2x35S:PHB-YFP, and slides coated with antibody that recognizes YFP, the authors found that 22% of mCherry and YFP spots colocalized. By contrast, overlapping images of different, random slide areas produced only 9% colocalization, as did analysis of noninteracting proteins; both values significantly differ from the 22% colocalization frequency of unfused mCherry and YFP (P < 10−24). Thus, these data confirm the interaction of these two proteins. The authors also examined the photobleaching steps for the complexes and found that PHB and ZPR3 form heterotetramers, rather than heterodimers, as previously thought.
So, is it time to consciously uncouple from your coimmunoprecipitation, bag your BiFC, and yank your yeast two-hybrid? On the one hand, SiMPull requires fluorescent labeling to detect proteins, and these fusions might interfere with protein-protein interactions. Proteins also are removed from their native context, so BiFC might be more useful for revealing the location of the interaction. SiMPull also requires some optimization to identify the proper protein concentrations and balance relative protein levels. On the other hand, despite these minor disadvantages, SiMPull provides a rapid, quantitative method to examine interactions between proteins (and a host of other biomolecules) and can directly determine the stoichiometries of immunoprecipitated complexes. Could the choice be more simple?
REFERENCES
Horstman, A., Tonaco, I.A., Boutilier, K., Immink, R.G. (2014). A cautionary note on the use of split-YFP/BiFC in plant protein-protein interaction studies. Int. J. Mol. Sci. 15: 9628–9643.
Husbands, A., Aggarwal, V., Ha, T., Timmermans, M.C.P. (2016). In planta single-molecule pull-down (SiMPull) reveals tetrameric stoichiometry of HD-ZIPIII:LITTLE ZIPPER complexes. Plant Cell 28: 1783–1794.
Jain, A., Liu, R., Ramani, B., Arauz, E., Ishitsuka, Y., Ragunathan, K., Park, J., Chen, J., Xiang, Y.K., Ha, T. (2011). Probing cellular protein complexes using single-molecule pull-down. Nature 473: 484–488.








Leave a Reply
Want to join the discussion?Feel free to contribute!