Plant Science Research Weekly: May 3, 2024
Review: The plant immune system: From discovery to deployment
 A review of the past 50 years of plant immunity by Jones, Staskawicz, and Dangl? Yes please! I particularly enjoy historical perspectives of a discipline, as they frame conceptual breakthroughs with the benefit of hindsight. As the article lays out, understanding the plant immune system benefitted greatly from the early advances in plant molecular biology and gene cloning, which allowed established concepts to be linked to proteins. Over a short period of time, several receptor proteins and pathogen effectors were identified, leading to the two-step model of cell-surface pattern-triggered immunity, backed up by intracellular effector-triggered immunity. This simple model has provided a structure that encompasses an extraordinary degree of nuance and subtlety, spurred in part by advances in structural analysis, sequencing, phylogenomics, and metagenomics. The authors conclude with a list of questions they would like to see solved in the next 20 years. Ultimately, it is the application of this knowledge to crop plants to enhance global food security that really matters, and this will be helped by CRISPR-based genome editing tools as well as an enlightened public who understand how today’s technology to support plant immunity connects to classical breeding; this article does an excellent job of bridging that gap. (Summary by Mary Williams @PlantTeaching) Cell 10.1016/j.cell.2024.03.045
A review of the past 50 years of plant immunity by Jones, Staskawicz, and Dangl? Yes please! I particularly enjoy historical perspectives of a discipline, as they frame conceptual breakthroughs with the benefit of hindsight. As the article lays out, understanding the plant immune system benefitted greatly from the early advances in plant molecular biology and gene cloning, which allowed established concepts to be linked to proteins. Over a short period of time, several receptor proteins and pathogen effectors were identified, leading to the two-step model of cell-surface pattern-triggered immunity, backed up by intracellular effector-triggered immunity. This simple model has provided a structure that encompasses an extraordinary degree of nuance and subtlety, spurred in part by advances in structural analysis, sequencing, phylogenomics, and metagenomics. The authors conclude with a list of questions they would like to see solved in the next 20 years. Ultimately, it is the application of this knowledge to crop plants to enhance global food security that really matters, and this will be helped by CRISPR-based genome editing tools as well as an enlightened public who understand how today’s technology to support plant immunity connects to classical breeding; this article does an excellent job of bridging that gap. (Summary by Mary Williams @PlantTeaching) Cell 10.1016/j.cell.2024.03.045
Perspective: Exposing belowground plant communication
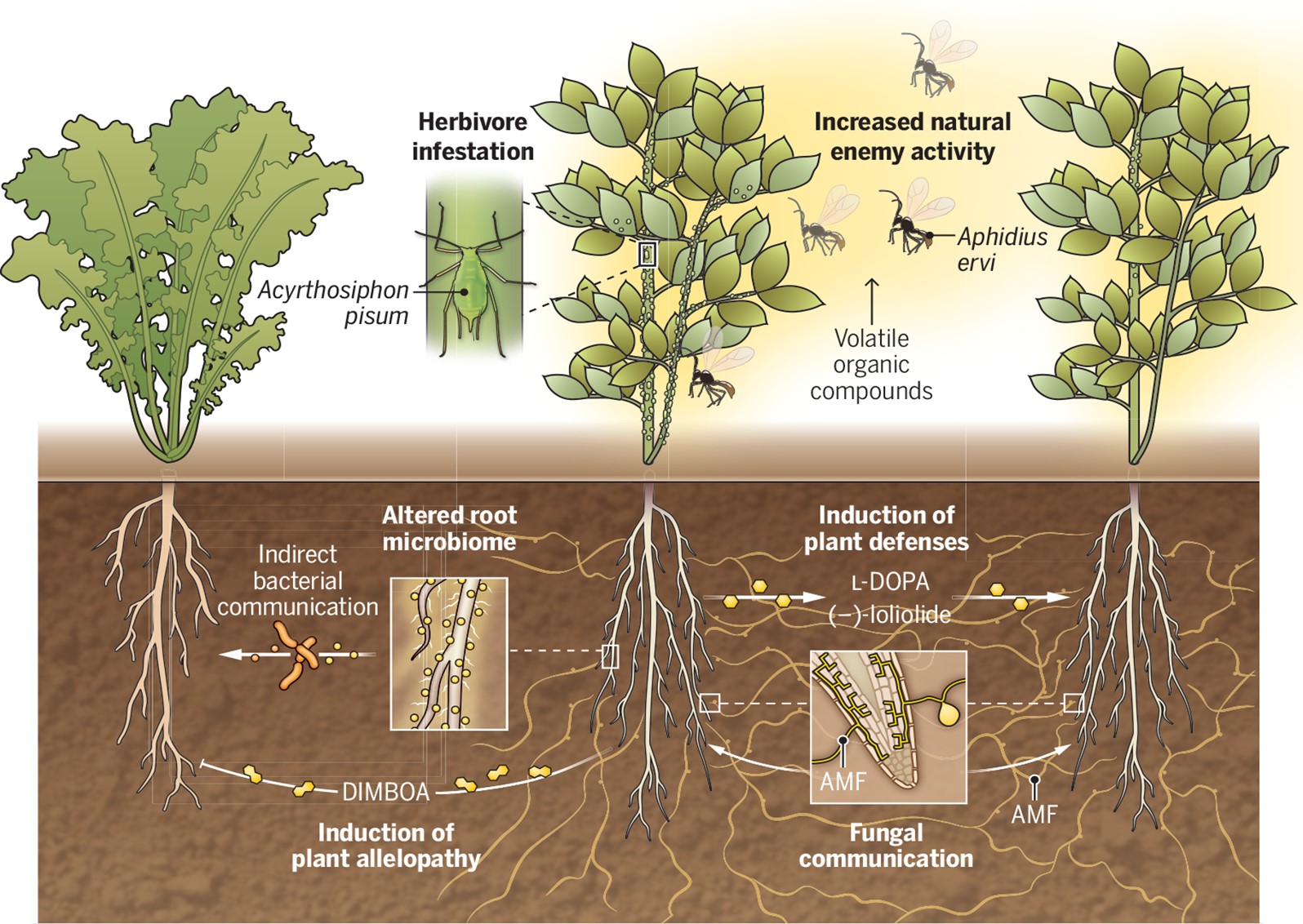 Plants possess a fascinating ability to communicate with each other through a complex system of chemical signals. Aboveground, they use airborne volatile signals to attract predatory insects, prime defenses in neighbors, facilitate nutrient transfer, and promote plant interactions. However, less is known about the chemical signaling that goes on underground. In this Perspective article, Guerrieri and Rasmann highlight what is known and unknown about belowground plant communication. As they suggest, soil matrix communication provides an efficient way to rapidly respond to biotic and abiotic stresses and can contribute to ecosystem processes. The authors provide examples of ways that root exudates communicate important information between organisms. Plants can also communicate through mycorrhizal networks to transfer available resources and signal between interconnected plants. However, understanding belowground plant communication calls for methodological advances, including sampling and characterizing root exudates in natural environments. Untargeted metabolomics and machine learning algorithms can detect molecules produced by roots under different stress conditions. Understanding and applying the potential of root exudates can be a game changer for sustainable agriculture and cost-effective ecosystem management. (Summary by Maneesh Lingwan, @LingwanManeesh) Science 10.1126/science.adk1412
Plants possess a fascinating ability to communicate with each other through a complex system of chemical signals. Aboveground, they use airborne volatile signals to attract predatory insects, prime defenses in neighbors, facilitate nutrient transfer, and promote plant interactions. However, less is known about the chemical signaling that goes on underground. In this Perspective article, Guerrieri and Rasmann highlight what is known and unknown about belowground plant communication. As they suggest, soil matrix communication provides an efficient way to rapidly respond to biotic and abiotic stresses and can contribute to ecosystem processes. The authors provide examples of ways that root exudates communicate important information between organisms. Plants can also communicate through mycorrhizal networks to transfer available resources and signal between interconnected plants. However, understanding belowground plant communication calls for methodological advances, including sampling and characterizing root exudates in natural environments. Untargeted metabolomics and machine learning algorithms can detect molecules produced by roots under different stress conditions. Understanding and applying the potential of root exudates can be a game changer for sustainable agriculture and cost-effective ecosystem management. (Summary by Maneesh Lingwan, @LingwanManeesh) Science 10.1126/science.adk1412
Review: The genomic route to tomato breeding: Past, present, and future
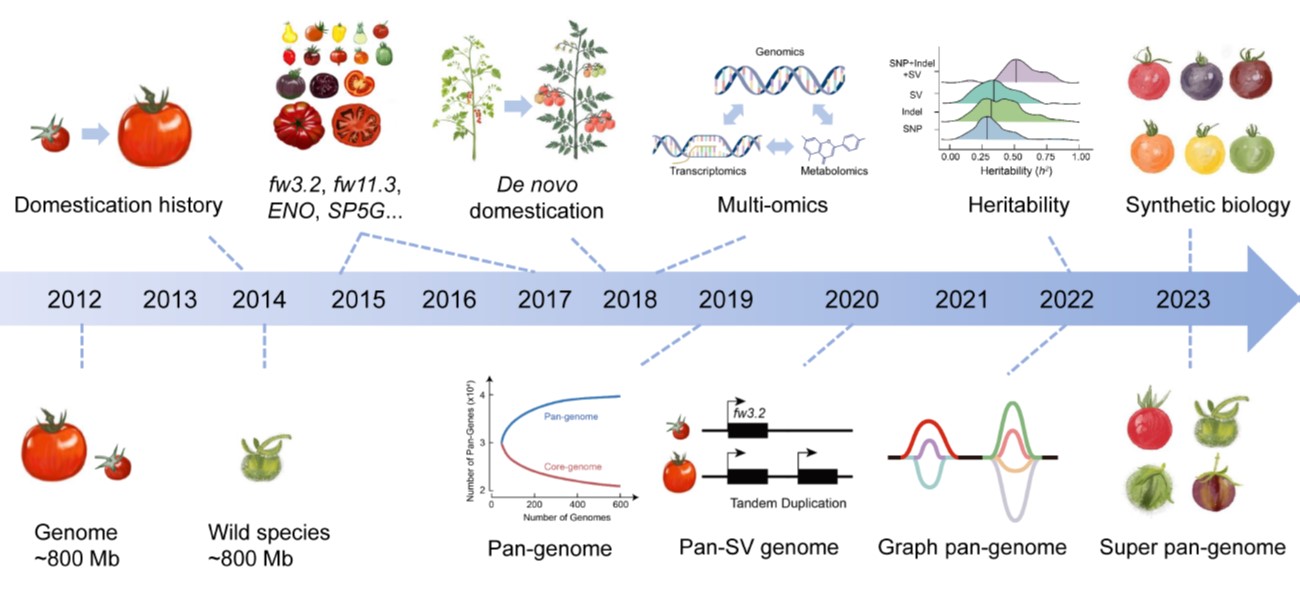 Widely and abundantly eaten tomatoes (Solanum lycopersicum) are delicious and nutritious, but the genetic diversity of cultivated tomatoes is quite narrow. In this review, Wang et al. give an overview of efforts to increase diversity, through introduction of genes from wild relatives and other approaches. Since the first tomato sequence was completed in 2012, countless sequences have been obtained from various cultivars and wild relatives, capturing the inter- and intra-species diversity. As a commercially important crop, tomatoes have been extensively analyzed for metabolic, developmental, resilience, and consumer preference traits, which have been mapped to genes and loci; there’s no shortage of data about tomatoes. Key traits that have been mapped pertain to flowering time, disease resistance, fruit weight, shape, ripening and quality. All this information provides the opportunity to produce exceptional varieties, but this is hindered by slow pace of conventional breeding. New technologies to accelerate breeding include haploid and male sterility breeding, heterosis, de novo domestication, gene editing, synthetic biology, and AI-assisted approaches. (Summary by Mary Williams @PlantTeaching) Plant Physiol. 10.1093/plphys/kiae248
Widely and abundantly eaten tomatoes (Solanum lycopersicum) are delicious and nutritious, but the genetic diversity of cultivated tomatoes is quite narrow. In this review, Wang et al. give an overview of efforts to increase diversity, through introduction of genes from wild relatives and other approaches. Since the first tomato sequence was completed in 2012, countless sequences have been obtained from various cultivars and wild relatives, capturing the inter- and intra-species diversity. As a commercially important crop, tomatoes have been extensively analyzed for metabolic, developmental, resilience, and consumer preference traits, which have been mapped to genes and loci; there’s no shortage of data about tomatoes. Key traits that have been mapped pertain to flowering time, disease resistance, fruit weight, shape, ripening and quality. All this information provides the opportunity to produce exceptional varieties, but this is hindered by slow pace of conventional breeding. New technologies to accelerate breeding include haploid and male sterility breeding, heterosis, de novo domestication, gene editing, synthetic biology, and AI-assisted approaches. (Summary by Mary Williams @PlantTeaching) Plant Physiol. 10.1093/plphys/kiae248
Phylogenomic insights into angiosperm evolution
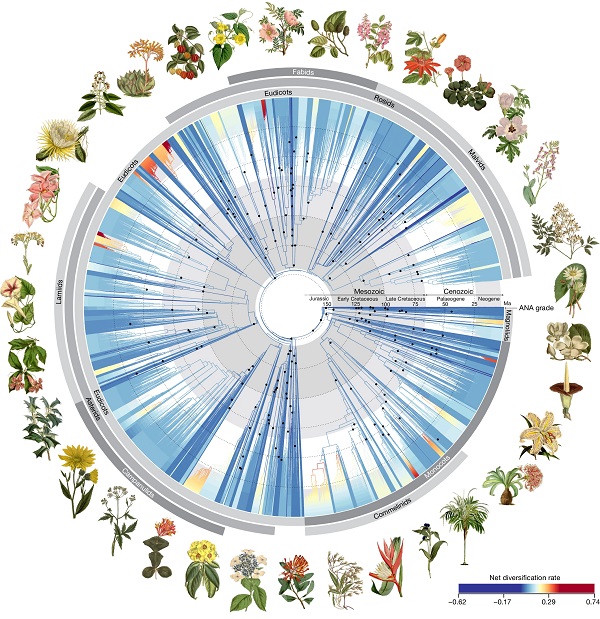 Low-resolution data can provide broad strokes but miss the details that come from greater information density. When striving to understand the multimillion-year evolutionary history of the angiosperms, more data certainly helps. Here, by focusing on a subset of 353 genes, Zuntini and Carruthers et al. present a phylogenomic tree that increases the number of represented angiosperm genera by fifteen-fold and includes all 416 families. The dataset encompasses 7,923 angiosperm genera, is time-calibrated through the analysis of a 200 fossil dataset, and enriched by samples from 163 herbaria in 48 countries. What does all this effort reveal? The results are beautifully presented in Figure 1 (available for download as a very high-resolution image, if, like me, you’d like to print it as a poster). Several clades have been reassigned, split, or lumped thanks to the new deeper dataset. The full dataset highlights two periods of exceptional diversification in the angiosperms; the first shortly after they emerged, during which about 80% of extant orders originated, and the second about 40 million years ago. The paper includes an Inclusion and Ethics statement, in which the authors acknowledge the people who collected and curated these samples and the complex histories underlying natural history collections. (Summary by Mary Williams @PlantTeaching) Nature 10.1038/s41586-024-07324-0
Low-resolution data can provide broad strokes but miss the details that come from greater information density. When striving to understand the multimillion-year evolutionary history of the angiosperms, more data certainly helps. Here, by focusing on a subset of 353 genes, Zuntini and Carruthers et al. present a phylogenomic tree that increases the number of represented angiosperm genera by fifteen-fold and includes all 416 families. The dataset encompasses 7,923 angiosperm genera, is time-calibrated through the analysis of a 200 fossil dataset, and enriched by samples from 163 herbaria in 48 countries. What does all this effort reveal? The results are beautifully presented in Figure 1 (available for download as a very high-resolution image, if, like me, you’d like to print it as a poster). Several clades have been reassigned, split, or lumped thanks to the new deeper dataset. The full dataset highlights two periods of exceptional diversification in the angiosperms; the first shortly after they emerged, during which about 80% of extant orders originated, and the second about 40 million years ago. The paper includes an Inclusion and Ethics statement, in which the authors acknowledge the people who collected and curated these samples and the complex histories underlying natural history collections. (Summary by Mary Williams @PlantTeaching) Nature 10.1038/s41586-024-07324-0
Genomics from bean to cup: New insights into the history of Arabica coffee diversification
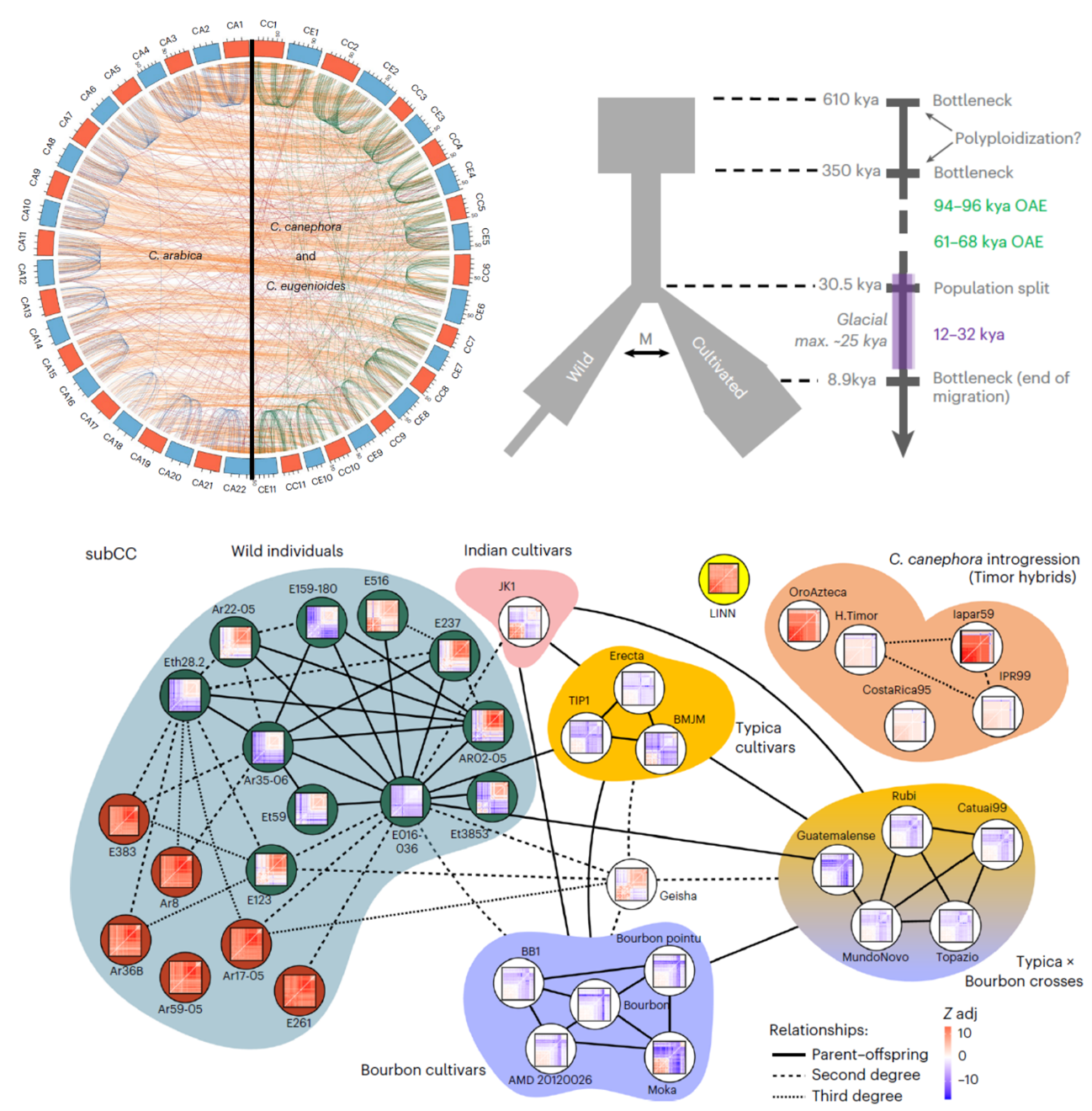 As one of the most traded commodities in the world, coffee has cultural and economic impact that spans continents. The main source of coffee beans, Coffea arabica (Arabica), is a polyploid species that resulted from the hybridization between diploid C. canephora (Robusta) and C. eugenioides (Eugenioides). Here, Salojärvi et al. produce chromosome-level assemblies of all three species, and reveal kinship and population dynamics estimates for 41 wild and cultivated coffee cultivars. No clear dominance expression pattern was observed between the Robusta- and Eugenioides-derived subgenomes of C. arabica. The overall low genetic diversity found in C. arabica, evidenced by its susceptibility to diseases such as leaf rust, is attributed to its relatively recent polyploid origin, estimated at 610-350 thousand years ago (ka). Population parameters point to a long bottleneck event (350-30.5 ka) ending with a more favourable climate for coffee growth and the split between today’s wild and cultivated populations. Migration across the split ended at ca. 9 ka, with wild populations alone facing another bottleneck, while cultivated Arabica spread to Yemen. In fact, Arabica cultivation is documented in Yemen in the 15th and 16th centuries, from where a handful of seeds made it to India; cultivation soon spread across Asian Dutch colonies. Some plants were brought to Europe and then spread by colonizers even more widely, notably from the island of Bourbon (present-day Réunion). Genomic data further documents the history of coffee: early Indian cultivar JK1 and later Typica and Bourbon cultivar groups are estimated to all share close kinship, reinforcing their ultimately Yemeni origin. Through natural hybridization with Robusta, a group of Timor Arabica cultivars are resistant to leaf rust. The Robusta introgression shared by resistant hybrids comprises blocks of candidate resistance genes that will be valuable tools in breeding strategies for leaf rust-resistant Arabica. (Summary by John Vilasboa @vilasjohn) Nat. Genet. 10.1038/s41588-024-01695-w
As one of the most traded commodities in the world, coffee has cultural and economic impact that spans continents. The main source of coffee beans, Coffea arabica (Arabica), is a polyploid species that resulted from the hybridization between diploid C. canephora (Robusta) and C. eugenioides (Eugenioides). Here, Salojärvi et al. produce chromosome-level assemblies of all three species, and reveal kinship and population dynamics estimates for 41 wild and cultivated coffee cultivars. No clear dominance expression pattern was observed between the Robusta- and Eugenioides-derived subgenomes of C. arabica. The overall low genetic diversity found in C. arabica, evidenced by its susceptibility to diseases such as leaf rust, is attributed to its relatively recent polyploid origin, estimated at 610-350 thousand years ago (ka). Population parameters point to a long bottleneck event (350-30.5 ka) ending with a more favourable climate for coffee growth and the split between today’s wild and cultivated populations. Migration across the split ended at ca. 9 ka, with wild populations alone facing another bottleneck, while cultivated Arabica spread to Yemen. In fact, Arabica cultivation is documented in Yemen in the 15th and 16th centuries, from where a handful of seeds made it to India; cultivation soon spread across Asian Dutch colonies. Some plants were brought to Europe and then spread by colonizers even more widely, notably from the island of Bourbon (present-day Réunion). Genomic data further documents the history of coffee: early Indian cultivar JK1 and later Typica and Bourbon cultivar groups are estimated to all share close kinship, reinforcing their ultimately Yemeni origin. Through natural hybridization with Robusta, a group of Timor Arabica cultivars are resistant to leaf rust. The Robusta introgression shared by resistant hybrids comprises blocks of candidate resistance genes that will be valuable tools in breeding strategies for leaf rust-resistant Arabica. (Summary by John Vilasboa @vilasjohn) Nat. Genet. 10.1038/s41588-024-01695-w
Genetic gains underpinning a little-known strawberry Green Revolution
 The domestication of cultivated strawberry (Fragaria × ananassa) traces back approximately 300 years, providing us with a relatively comprehensive genealogy of this artificial hybrid species. Strawberry yield in the US has increased by 2,755% since the 1960’s, largely owing to the California strawberry Green Revolution. Through an analysis of California strawberry germplasm, researchers have pinpointed significant factors contributing to this remarkable increase. One crucial contributor was the introduction of photoperiod-insensitive, PERPETUAL FLOWERING hybrids during the 1970s, which substantially enhanced yield potential. Additionally, the adoption of methyl bromide fumigation played a pivotal role by significantly reducing yield losses attributed to diseases. However, the widespread use of fumigation has led to a decline in resistance among contemporary strawberry accessions to soil-borne pathogens such as Verticillium wilt and Phytophthora crown rot. This poses a potential threat to strawberry production in the US, especially with the phasing out of methyl bromide. (Summary by Villő Bernád) Nature Comms. 10.1038/s41467-024-46421-6
The domestication of cultivated strawberry (Fragaria × ananassa) traces back approximately 300 years, providing us with a relatively comprehensive genealogy of this artificial hybrid species. Strawberry yield in the US has increased by 2,755% since the 1960’s, largely owing to the California strawberry Green Revolution. Through an analysis of California strawberry germplasm, researchers have pinpointed significant factors contributing to this remarkable increase. One crucial contributor was the introduction of photoperiod-insensitive, PERPETUAL FLOWERING hybrids during the 1970s, which substantially enhanced yield potential. Additionally, the adoption of methyl bromide fumigation played a pivotal role by significantly reducing yield losses attributed to diseases. However, the widespread use of fumigation has led to a decline in resistance among contemporary strawberry accessions to soil-borne pathogens such as Verticillium wilt and Phytophthora crown rot. This poses a potential threat to strawberry production in the US, especially with the phasing out of methyl bromide. (Summary by Villő Bernád) Nature Comms. 10.1038/s41467-024-46421-6
Enhanced super-resolution ribosome profiling unveils pervasive translation of upstream ORFs and small ORFs in Arabidopsis
 In recent years, the discovery that the translational regulation of many mRNAs is influenced by small upstream open reading frames (uORFs) has highlighted the importance of precisely identifying gene structures and translated ORFs to understand gene functions and cellular behavior. Using Arabidopsis, Wu et al. advanced the precision of ribosome profiling, in which RNA sequences bound by ribosomes are identified, revealing 7,751 unconventional translation occurrences. Subsequent validation via proteomic scrutiny affirmed the production of stable proteins from the identified sites. Noteworthy instances of active translation were observed in small interfering RNAs and microRNAs. A novel methodology was developed to discern short upstream ORFs (uORFs), showcasing distinct translational repression patterns. Moreover, the recognition of 594 uORFs regulated through alternative splicing, and their involvement in crucial pathways, underscores the comprehensive nature of this investigation in elucidating the mechanisms governing gene expression regulation in Arabidopsis. (Summary by Yueh Cho @YuehCho1984) Plant Cell. 10.1093/plcell/koad290.
In recent years, the discovery that the translational regulation of many mRNAs is influenced by small upstream open reading frames (uORFs) has highlighted the importance of precisely identifying gene structures and translated ORFs to understand gene functions and cellular behavior. Using Arabidopsis, Wu et al. advanced the precision of ribosome profiling, in which RNA sequences bound by ribosomes are identified, revealing 7,751 unconventional translation occurrences. Subsequent validation via proteomic scrutiny affirmed the production of stable proteins from the identified sites. Noteworthy instances of active translation were observed in small interfering RNAs and microRNAs. A novel methodology was developed to discern short upstream ORFs (uORFs), showcasing distinct translational repression patterns. Moreover, the recognition of 594 uORFs regulated through alternative splicing, and their involvement in crucial pathways, underscores the comprehensive nature of this investigation in elucidating the mechanisms governing gene expression regulation in Arabidopsis. (Summary by Yueh Cho @YuehCho1984) Plant Cell. 10.1093/plcell/koad290.
Heat stress promotes Arabidopsis AGO1 phase separation and association with stress granule components
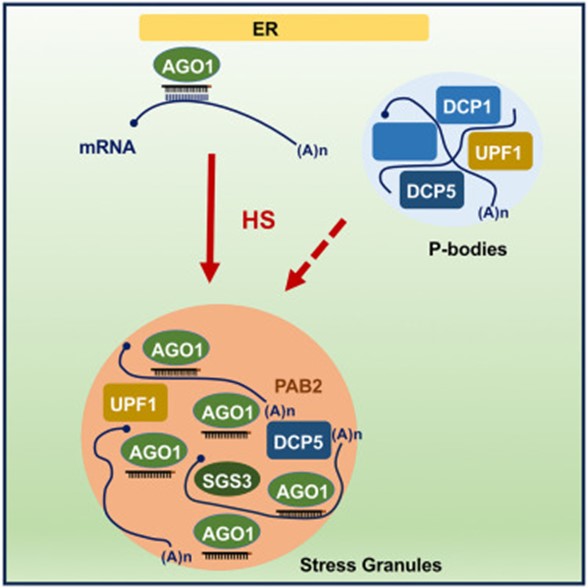 A new article by Blagojevic, Baldrich, and Schiaffini et al. reveals that Arabidopsis ARGONAUTE1 (AGO1) protein, a pivotal agent in miRNA and siRNA-mediated gene silencing associated with the rough endoplasmic reticulum, dynamically localizes within stress granule components during heat stress (HS). This relocalization does not require SUPPRESSOR OF GENE SILENCING 3 (SGS3) and includes interactions with P-bodies and RNA decay components, forming a unique HS-induced AGO1 interactome. Moreover, even a short period of HS at 37°C results in only limited changes to small RNA (sRNA) accumulation and their loading into AGO1, indicating the resilience of AGO1’s silencing capabilities under HS. A significant finding is that AGO1 undergoes liquid-liquid phase separation through its N-terminal Poly-Q domain, which may contribute to its protective aggregation in cytosolic condensates. This study enhances our understanding of the molecular responses to HS in plants, highlighting the robust nature of AGO1 and its gene regulatory functions during environmental stress. (Summary by Yueh Cho @YuehCho1984) iScience. 10.1016/j.isci.2024.109151
A new article by Blagojevic, Baldrich, and Schiaffini et al. reveals that Arabidopsis ARGONAUTE1 (AGO1) protein, a pivotal agent in miRNA and siRNA-mediated gene silencing associated with the rough endoplasmic reticulum, dynamically localizes within stress granule components during heat stress (HS). This relocalization does not require SUPPRESSOR OF GENE SILENCING 3 (SGS3) and includes interactions with P-bodies and RNA decay components, forming a unique HS-induced AGO1 interactome. Moreover, even a short period of HS at 37°C results in only limited changes to small RNA (sRNA) accumulation and their loading into AGO1, indicating the resilience of AGO1’s silencing capabilities under HS. A significant finding is that AGO1 undergoes liquid-liquid phase separation through its N-terminal Poly-Q domain, which may contribute to its protective aggregation in cytosolic condensates. This study enhances our understanding of the molecular responses to HS in plants, highlighting the robust nature of AGO1 and its gene regulatory functions during environmental stress. (Summary by Yueh Cho @YuehCho1984) iScience. 10.1016/j.isci.2024.109151
Conjugation of ATG8 to vacuolar membranes as a response to cell wall damage
 ATG8 is a well-characterized protein involved in autophagy that binds to the double-membrane enclosed phagophore. In a new preprint, Julian et al. explore their finding that ATG8 binds to the single-membrane enclosed vacuolar membrane (tonoplast). They observed that this binding is enhanced by treatments that weaken the cell wall, such as inhibition of cellulose synthase. To identify its function, they generated plants defective in conjugation of ATG8 to single membranes (ΔCASM), which blocks this specific interaction but does not affect autophagy more broadly. ΔCASM plants show defects in tonoplast and vacuolar integrity and are more sensitive to cell-wall damage. The authors propose a vacuolar quality control (VQC) pathway in which cell wall damage triggers a change in turgor pressure and vacuolar pH which leads to ATG8 recruitment and membrane remodeling to prevent tonoplast lysis in conditions where cell wall integrity is damaged. This work has identified a new quality control pathway for vacuoles and links ATG8 with cell integrity sensing. (Summary by Mary Williams @PlantTeaching) bioRxiv 10.1101/2024.04.21.590262
ATG8 is a well-characterized protein involved in autophagy that binds to the double-membrane enclosed phagophore. In a new preprint, Julian et al. explore their finding that ATG8 binds to the single-membrane enclosed vacuolar membrane (tonoplast). They observed that this binding is enhanced by treatments that weaken the cell wall, such as inhibition of cellulose synthase. To identify its function, they generated plants defective in conjugation of ATG8 to single membranes (ΔCASM), which blocks this specific interaction but does not affect autophagy more broadly. ΔCASM plants show defects in tonoplast and vacuolar integrity and are more sensitive to cell-wall damage. The authors propose a vacuolar quality control (VQC) pathway in which cell wall damage triggers a change in turgor pressure and vacuolar pH which leads to ATG8 recruitment and membrane remodeling to prevent tonoplast lysis in conditions where cell wall integrity is damaged. This work has identified a new quality control pathway for vacuoles and links ATG8 with cell integrity sensing. (Summary by Mary Williams @PlantTeaching) bioRxiv 10.1101/2024.04.21.590262
Photoperiod-insensitive flowering is associated with the FT gene in hemp
 Cannabis sativa, also known as hemp or marijuana, is a widely cultivated plant for a variety of reasons. It is perhaps best known as a producer of an intoxicating chemical, THC, produced by glandular trichomes on female flowers, but low-THC producers (hemp) are cultivated for fibers. Flowering time affects both THC and fiber production, and various cultivars have a wide range of flowering-time responses. Here, Dowling et al. explored the genetic basis for photoperiod insensitive flowering time in the cultivar FINOLA. They identified a candidate locus, Autoflower2, that includes a tandem duplication of CsFT1, an ortholog of FLOWERING LOCUS T (FT). Interestingly, in photoperiod sensitive cultivars, this gene exists in a single copy. Furthermore, the expression of the genes in FINOLA is photoperiod insensitive, whereas it is only expressed in flower-inducing conditions in photoperiod sensitive cultivars. The authors surveyed a population of cultivars and found a correlation with the FINOLA-type CsFT1 locus and photoperiod insensitivity, although other genes are likely to contribute as well. (Summary by Mary Williams @PlantTeaching) Plant J. 10.1111/tpj.16769
Cannabis sativa, also known as hemp or marijuana, is a widely cultivated plant for a variety of reasons. It is perhaps best known as a producer of an intoxicating chemical, THC, produced by glandular trichomes on female flowers, but low-THC producers (hemp) are cultivated for fibers. Flowering time affects both THC and fiber production, and various cultivars have a wide range of flowering-time responses. Here, Dowling et al. explored the genetic basis for photoperiod insensitive flowering time in the cultivar FINOLA. They identified a candidate locus, Autoflower2, that includes a tandem duplication of CsFT1, an ortholog of FLOWERING LOCUS T (FT). Interestingly, in photoperiod sensitive cultivars, this gene exists in a single copy. Furthermore, the expression of the genes in FINOLA is photoperiod insensitive, whereas it is only expressed in flower-inducing conditions in photoperiod sensitive cultivars. The authors surveyed a population of cultivars and found a correlation with the FINOLA-type CsFT1 locus and photoperiod insensitivity, although other genes are likely to contribute as well. (Summary by Mary Williams @PlantTeaching) Plant J. 10.1111/tpj.16769
Significant floral diversity independent of pollinator change in ‘Buzz-Bee’ pollinated Melastomataceae
 The concept of pollination syndromes posits that floral diversity arises from shifts among pollinator groups, yet their predictive accuracy for pollinator identification is debated. Kopper et al. leveraged machine learning, utilizing 44 floral traits from 252 species with known pollinator interactions, to predict pollinators for 159 species lacking empirical data. Concurrently, they used multivariate and phylogenetic analyses to examine the link between pollinator shifts and floral disparity within Melastomataceae, a diverse family of flowering plants found mainly in tropical regions. The findings confirmed four distinct pollination syndromes and revealed that, while pollinator transitions contribute to floral diversity, the greatest disparity was observed in the species-rich ‘buzz-bee’ syndrome, suggesting that significant floral variation can develop without pollinator shifts. Buzz-pollinated flowers release pollen only when bees apply specific vibrations to the flower, and in many families buzz-pollinated species show similar flower morphologies. Additionally, species-poor clades and certain regions also significantly contributed to floral diversity. Overall, their study underscores the potential of machine learning in evaluating pollination syndromes and predicting pollinators where direct evidence is unavailable. (Summary by Yueh Cho @YuehCho1984) New Phytologist. 10.1111/nph.19735
The concept of pollination syndromes posits that floral diversity arises from shifts among pollinator groups, yet their predictive accuracy for pollinator identification is debated. Kopper et al. leveraged machine learning, utilizing 44 floral traits from 252 species with known pollinator interactions, to predict pollinators for 159 species lacking empirical data. Concurrently, they used multivariate and phylogenetic analyses to examine the link between pollinator shifts and floral disparity within Melastomataceae, a diverse family of flowering plants found mainly in tropical regions. The findings confirmed four distinct pollination syndromes and revealed that, while pollinator transitions contribute to floral diversity, the greatest disparity was observed in the species-rich ‘buzz-bee’ syndrome, suggesting that significant floral variation can develop without pollinator shifts. Buzz-pollinated flowers release pollen only when bees apply specific vibrations to the flower, and in many families buzz-pollinated species show similar flower morphologies. Additionally, species-poor clades and certain regions also significantly contributed to floral diversity. Overall, their study underscores the potential of machine learning in evaluating pollination syndromes and predicting pollinators where direct evidence is unavailable. (Summary by Yueh Cho @YuehCho1984) New Phytologist. 10.1111/nph.19735
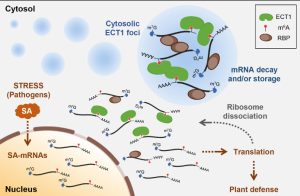 Findings: Among the 13 YTH family proteins in Arabidopsis, we identified EVOLUTIONARILY CONSERVED C-TERMINAL REGION1 (ECT1) as a crucial player in SA-mediated stress responses. Loss of ECT1 enhances SA-primed growth retardation and cell death. ECT1 undergoes liquid-liquid phase separation, forming cytosolic biomolecular condensates such as processing bodies and stress granules, sites for decay and/or storage of non-translating mRNAs, in response to SA or bacterial pathogens. It sequesters SA-induced m6A modification-prone mRNAs (SA-m6A-mRNAs), facilitating their decay in cytosolic biomolecular condensates, thus compromising SA-mediated stress responses. Arabidopsis plants overexpressing ECT1 are more susceptible to bacterial pathogens.
Findings: Among the 13 YTH family proteins in Arabidopsis, we identified EVOLUTIONARILY CONSERVED C-TERMINAL REGION1 (ECT1) as a crucial player in SA-mediated stress responses. Loss of ECT1 enhances SA-primed growth retardation and cell death. ECT1 undergoes liquid-liquid phase separation, forming cytosolic biomolecular condensates such as processing bodies and stress granules, sites for decay and/or storage of non-translating mRNAs, in response to SA or bacterial pathogens. It sequesters SA-induced m6A modification-prone mRNAs (SA-m6A-mRNAs), facilitating their decay in cytosolic biomolecular condensates, thus compromising SA-mediated stress responses. Arabidopsis plants overexpressing ECT1 are more susceptible to bacterial pathogens.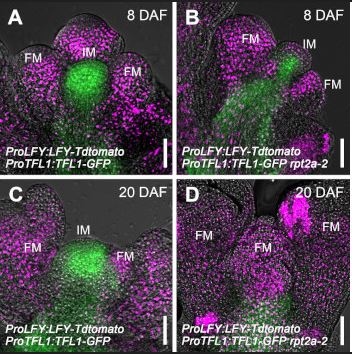 Findings: We present insights into the mechanisms that fine-tune the activity of TFL1, a key regulator of indeterminate inflorescence growth in Arabidopsis. Our findings demonstrate that TFL1 levels in the IM are regulated by DNA methylation mediated by the DNA methyltransferase MET1. Low MET1 levels are maintained through proteolysis via the 26S proteasome subunit RPT2a, which keeps methylation at the TFL1 promoter low, thus promoting IM indeterminacy and indeterminate inflorescence architecture. Conversely, when RPT2a-mediated proteolysis of MET1 is hindered, TFL1 expression levels decrease, leading to a determinate zigzag-shaped inflorescence architecture. Our findings propose an integrative model that explains how proteolytic activity balances the epigenetic regulation of TFL1 expression, ultimately leading to indeterminate inflorescence architecture in Arabidopsis.
Findings: We present insights into the mechanisms that fine-tune the activity of TFL1, a key regulator of indeterminate inflorescence growth in Arabidopsis. Our findings demonstrate that TFL1 levels in the IM are regulated by DNA methylation mediated by the DNA methyltransferase MET1. Low MET1 levels are maintained through proteolysis via the 26S proteasome subunit RPT2a, which keeps methylation at the TFL1 promoter low, thus promoting IM indeterminacy and indeterminate inflorescence architecture. Conversely, when RPT2a-mediated proteolysis of MET1 is hindered, TFL1 expression levels decrease, leading to a determinate zigzag-shaped inflorescence architecture. Our findings propose an integrative model that explains how proteolytic activity balances the epigenetic regulation of TFL1 expression, ultimately leading to indeterminate inflorescence architecture in Arabidopsis.