Review: Guidelines for studying and naming plant plasma-membrane domains
 Numerous studies have highlighted the critical importance of plasma membrane heterogeneities in regulating cell functions, leading to a proliferation of overlapping and contradictory terminologies. Here, Jaillais and others in the field propose a new system of nomenclature. It really is a must-read for anyone interested in cell biology, so here I’ll just report two key recommendations. First, they propose a clear distinction between nanodomains and polar domains, largely dependent on scale. Nanodomains are small (<1 μm) molecular assemblies in the membrane plane. Polar domains are larger, site-specific accumulations of membrane molecules occurring at the cellular level, often associated with cell-level processes such as vesicular transport or a polarized cytoskeleton. Second, the authors provide a thoughtful overview of the driver/ client relationship in membrane domains, as well as guidelines for studying membrane domains. They conclude, “With the progress of fluorescence microscopy techniques with ever-increasing spatiotemporal resolution, the study of membrane domains has become a major focus of plant cell biology, and we anticipate that this interest in the community will continue to grow in the coming years.” (Summary by Mary Williams @PlantTeaching) Nature Plants 10.1038/s41477-024-01742-8
Numerous studies have highlighted the critical importance of plasma membrane heterogeneities in regulating cell functions, leading to a proliferation of overlapping and contradictory terminologies. Here, Jaillais and others in the field propose a new system of nomenclature. It really is a must-read for anyone interested in cell biology, so here I’ll just report two key recommendations. First, they propose a clear distinction between nanodomains and polar domains, largely dependent on scale. Nanodomains are small (<1 μm) molecular assemblies in the membrane plane. Polar domains are larger, site-specific accumulations of membrane molecules occurring at the cellular level, often associated with cell-level processes such as vesicular transport or a polarized cytoskeleton. Second, the authors provide a thoughtful overview of the driver/ client relationship in membrane domains, as well as guidelines for studying membrane domains. They conclude, “With the progress of fluorescence microscopy techniques with ever-increasing spatiotemporal resolution, the study of membrane domains has become a major focus of plant cell biology, and we anticipate that this interest in the community will continue to grow in the coming years.” (Summary by Mary Williams @PlantTeaching) Nature Plants 10.1038/s41477-024-01742-8
Mechanism of auxin-dependent gene regulation through composite auxin response elements
 Auxin signaling influences plant growth and development by controlling gene expression, often through binding of transcription factors from the Auxin Response Factor (ARF) family to ARF elements (AuxRE) present in the promoters of auxin-responsive genes. However, given that auxin signaling produces many varied effects on growth, it’s not clear how diverse responses are achieved with only a limited set of ARF transcription factors. One possibility is that sequences adjacent to the AuxRE, forming composite auxin response elements, influence the response by facilitating interactions between the ARF and other transcription factors. Novikova et al. investigated how these composite AuxRE’s contribute to regulation of auxin-responsive genes using computational, functional genomics, and classic genetic analyses. They demonstrated that ARF transcription factors interact with an array of binding partners to regulate composite AuxRE’s and fine-tune gene expression. They explored the role of a composite AuxRE module present in the promoter of the auxin-responsive INDOLE-3-ACETIC ACID INDUCIBLE 30 (IAA30) gene and demonstrated that stacking of composite AuxRE’s is important for IAA30‘s function in root development. Composite AuxRE modules may provide a mechanism for nuanced control of auxin signaling and are likely important for mediating auxin growth responses during development and in stressful environments. (Summary by Alicia Quinn @AliciaQuinnSci). bioRxiv 10.1101/2024.07.16.603724
Auxin signaling influences plant growth and development by controlling gene expression, often through binding of transcription factors from the Auxin Response Factor (ARF) family to ARF elements (AuxRE) present in the promoters of auxin-responsive genes. However, given that auxin signaling produces many varied effects on growth, it’s not clear how diverse responses are achieved with only a limited set of ARF transcription factors. One possibility is that sequences adjacent to the AuxRE, forming composite auxin response elements, influence the response by facilitating interactions between the ARF and other transcription factors. Novikova et al. investigated how these composite AuxRE’s contribute to regulation of auxin-responsive genes using computational, functional genomics, and classic genetic analyses. They demonstrated that ARF transcription factors interact with an array of binding partners to regulate composite AuxRE’s and fine-tune gene expression. They explored the role of a composite AuxRE module present in the promoter of the auxin-responsive INDOLE-3-ACETIC ACID INDUCIBLE 30 (IAA30) gene and demonstrated that stacking of composite AuxRE’s is important for IAA30‘s function in root development. Composite AuxRE modules may provide a mechanism for nuanced control of auxin signaling and are likely important for mediating auxin growth responses during development and in stressful environments. (Summary by Alicia Quinn @AliciaQuinnSci). bioRxiv 10.1101/2024.07.16.603724
UBIQUITIN-SPECIFIC PROTEASE (UBP14) interacts with HY5 to promote photomorphogenesis under dark-to-light conditions
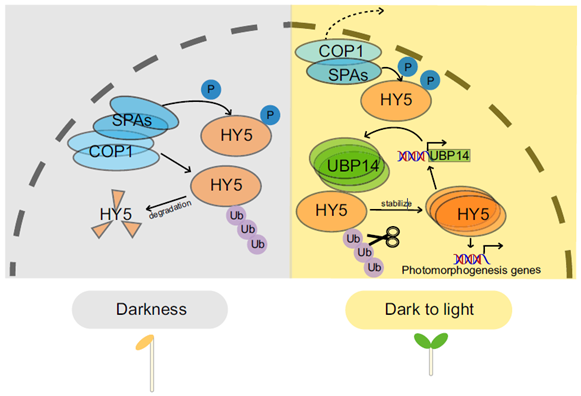
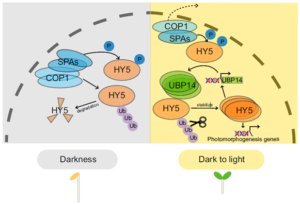 ELONGATED HYPOCOTYL5 (HY5) is a transcription factor that regulates about one-third of Arabidopsis genes, affecting growth and development of seedlings through light and hormone signaling. UBIQUITIN-SPECIFIC PROTEASE (UBP14) is a deubiquitinating enzyme that removes ubiquitin from substrate proteins. In a new study, Fang et al. performed bi-molecular fluorescence complementation, and yeast two hybrid, pull down, and co-immunoprecipitation assays to validate the interaction of UBP14 and HY5 in vitro and in vivo. Their experiments show that UBP14 is involved in stabilizing the protein expression of HY5 under dark-to-light conditions, which is essential for photomorphogenesis (promoting cotyledon opening and inhibiting hypocotyl growth). On the contrary, the absence of UBP14 leads to a slower accumulation of HY5 protein. UBP14 preferentially stabilizes the non-phosphorylated form of HY5 which is more active in binding other downstream genes compared to the phosphorylated form. HY5 can directly bind to two G-box elements present near the transcription start site in the promoter of UBP14 and promote its positive feedback, making itself more stable under light conditions. This discovery provides an excellent example of how a deubiquitinating enzyme contributes to cotyledon opening. (Summary by Asif Ali @pbgasifkalas) PNAS. 10.1073/pnas.2404883121
ELONGATED HYPOCOTYL5 (HY5) is a transcription factor that regulates about one-third of Arabidopsis genes, affecting growth and development of seedlings through light and hormone signaling. UBIQUITIN-SPECIFIC PROTEASE (UBP14) is a deubiquitinating enzyme that removes ubiquitin from substrate proteins. In a new study, Fang et al. performed bi-molecular fluorescence complementation, and yeast two hybrid, pull down, and co-immunoprecipitation assays to validate the interaction of UBP14 and HY5 in vitro and in vivo. Their experiments show that UBP14 is involved in stabilizing the protein expression of HY5 under dark-to-light conditions, which is essential for photomorphogenesis (promoting cotyledon opening and inhibiting hypocotyl growth). On the contrary, the absence of UBP14 leads to a slower accumulation of HY5 protein. UBP14 preferentially stabilizes the non-phosphorylated form of HY5 which is more active in binding other downstream genes compared to the phosphorylated form. HY5 can directly bind to two G-box elements present near the transcription start site in the promoter of UBP14 and promote its positive feedback, making itself more stable under light conditions. This discovery provides an excellent example of how a deubiquitinating enzyme contributes to cotyledon opening. (Summary by Asif Ali @pbgasifkalas) PNAS. 10.1073/pnas.2404883121
APETALA2 coordinates shoot apical meristem shape and identity during floral transition
 The study by Bertran Garcia de Olalla et al. investigates the role of the APETALA2 (AP2) transcription factor in Arabidopsis, particularly its influence on the shoot apical meristem (SAM) during the transition from vegetative to reproductive growth. AP2 is essential for the rapid increase in SAM height and width, which is associated with changes in the central zone and organizing center. As the floral transition progresses, AP2 expression decreases, regulated in part by the gene SUPPRESSOR OF OVEREXPRESSION OF CONSTANS1 (SOC1) and the microRNA miR172. The study highlights the mutual repression between AP2 and SOC1, which plays a crucial role in synchronizing changes in SAM morphology with the plant’s transition to flower production. AP2’s presence in the SAM is essential for establishing and maintaining the characteristic domed shape of the SAM during the floral transition. Moreover, prolonging AP2 expression beyond its typical repression point results in wider SAMs, whereas SOC1 not only promotes the floral transition but also modulates SAM size by reducing both its width and height when overexpressed. The study also shows that AP2 influences cell number and size within the SAM, further contributing to its overall morphology. These findings reveal the gene networks that control the timing and execution of flowering in response to environmental signals. (Summary by Amarachi Ezeoke) Nature Comms. 10.1038/s41467-024-51341-6
The study by Bertran Garcia de Olalla et al. investigates the role of the APETALA2 (AP2) transcription factor in Arabidopsis, particularly its influence on the shoot apical meristem (SAM) during the transition from vegetative to reproductive growth. AP2 is essential for the rapid increase in SAM height and width, which is associated with changes in the central zone and organizing center. As the floral transition progresses, AP2 expression decreases, regulated in part by the gene SUPPRESSOR OF OVEREXPRESSION OF CONSTANS1 (SOC1) and the microRNA miR172. The study highlights the mutual repression between AP2 and SOC1, which plays a crucial role in synchronizing changes in SAM morphology with the plant’s transition to flower production. AP2’s presence in the SAM is essential for establishing and maintaining the characteristic domed shape of the SAM during the floral transition. Moreover, prolonging AP2 expression beyond its typical repression point results in wider SAMs, whereas SOC1 not only promotes the floral transition but also modulates SAM size by reducing both its width and height when overexpressed. The study also shows that AP2 influences cell number and size within the SAM, further contributing to its overall morphology. These findings reveal the gene networks that control the timing and execution of flowering in response to environmental signals. (Summary by Amarachi Ezeoke) Nature Comms. 10.1038/s41467-024-51341-6
Single cell transcriptomics aids gene discovery of complex natural product biosynthesis
 From an ancient Greek cure-all to a modern treatment for mild depression, Hypericum perforatum (St. John’s wort) is a fascinating weed. Its leaves and flowers produce hyperforin, a metabolite derived from the isoprenoid pathway, which acts as a serotonin reuptake inhibitor. Despite partial genome and transcriptome data, the later steps in its biosynthesis pathway remain unclear. In a recent publication in Molecular Plant, Wu and coauthors aimed to uncover these missing steps. They used whole genome sequencing together with single-cell RNA sequencing (scRNA-seq) of leaf- and flower-derived protoplasts to identify a cell cluster with high expression of known biosynthesis genes, denominated “Hyper cells”. GO analysis revealed four putative genes expressed in these Hyper cells for the elusive last steps of the pathway. These candidates, called HpPT1 to 4, were cloned and reconstituted in yeast, where they were shown to catalyze the different prenylation reactions, which was further validated using a transient expression system in planta. Their study explains previous difficulties in gene discovery, as candidate genes were likely masked using bulk RNA-sequencing. In conclusion, the authors provide a complete elucidation of the hyperforin biosynthesis pathway and successful production of hyperforin in yeast which offers tremendous possibilities for the optimization of hyperforin heterologous cell factories. In addition, this study nicely highlights the power of scRNA-seq in overcoming challenges in gene discovery, apart from its classical applications. (Summary by Thomas Depaepe @thdpaepe). Molecular Plant 10.1016/j.molp.2024.08.003.
From an ancient Greek cure-all to a modern treatment for mild depression, Hypericum perforatum (St. John’s wort) is a fascinating weed. Its leaves and flowers produce hyperforin, a metabolite derived from the isoprenoid pathway, which acts as a serotonin reuptake inhibitor. Despite partial genome and transcriptome data, the later steps in its biosynthesis pathway remain unclear. In a recent publication in Molecular Plant, Wu and coauthors aimed to uncover these missing steps. They used whole genome sequencing together with single-cell RNA sequencing (scRNA-seq) of leaf- and flower-derived protoplasts to identify a cell cluster with high expression of known biosynthesis genes, denominated “Hyper cells”. GO analysis revealed four putative genes expressed in these Hyper cells for the elusive last steps of the pathway. These candidates, called HpPT1 to 4, were cloned and reconstituted in yeast, where they were shown to catalyze the different prenylation reactions, which was further validated using a transient expression system in planta. Their study explains previous difficulties in gene discovery, as candidate genes were likely masked using bulk RNA-sequencing. In conclusion, the authors provide a complete elucidation of the hyperforin biosynthesis pathway and successful production of hyperforin in yeast which offers tremendous possibilities for the optimization of hyperforin heterologous cell factories. In addition, this study nicely highlights the power of scRNA-seq in overcoming challenges in gene discovery, apart from its classical applications. (Summary by Thomas Depaepe @thdpaepe). Molecular Plant 10.1016/j.molp.2024.08.003.
Comparative transcriptomics in ferns provide a framework for their unique evolutionary path
 Ferns are important and diverse land plants but are also known for their exceptionally large genomes. A new study by Ali et al. presents an extensive analysis of fern genomics through RNA-sequencing of 22 representative fern species. The study identified 18 whole-genome duplications across different fern lineages, contributing to their high chromosome numbers and species diversity. The analysis of fern gene functions shows that over 50% of gene families are fern-specific, indicating novel, unexplored functions that differentiate them from other land plants. The study also highlights the absence of several genes crucial for hormonal signaling, defense, and development found in angiosperms, reflecting ferns’ unique strategies for growth and survival. Additionally, the research revealed the presence of an unusual sugar, 2-O-methyl-D-glucopyranose, in certain fern species, suggesting a divergent evolutionary path in cell wall biochemistry. An investigation into fern cell walls identifies the presence of mixed-linkage glucans and variations in polysaccharide composition, underscoring the complexity and diversity of fern cell wall structures. (Summary by Amarachi Ezeoke) bioRxiv https://doi.org/10.1101/2024.08.27.609851
Ferns are important and diverse land plants but are also known for their exceptionally large genomes. A new study by Ali et al. presents an extensive analysis of fern genomics through RNA-sequencing of 22 representative fern species. The study identified 18 whole-genome duplications across different fern lineages, contributing to their high chromosome numbers and species diversity. The analysis of fern gene functions shows that over 50% of gene families are fern-specific, indicating novel, unexplored functions that differentiate them from other land plants. The study also highlights the absence of several genes crucial for hormonal signaling, defense, and development found in angiosperms, reflecting ferns’ unique strategies for growth and survival. Additionally, the research revealed the presence of an unusual sugar, 2-O-methyl-D-glucopyranose, in certain fern species, suggesting a divergent evolutionary path in cell wall biochemistry. An investigation into fern cell walls identifies the presence of mixed-linkage glucans and variations in polysaccharide composition, underscoring the complexity and diversity of fern cell wall structures. (Summary by Amarachi Ezeoke) bioRxiv https://doi.org/10.1101/2024.08.27.609851
Phylogenomics of rubber trees sheds light on latex production
 Natural rubber, primarily derived from Hevea brasiliensis, is an essential global resource, but its production threatened by environmental changes and pest pressures. Fang et al. examined genome assemblies from eight high-quality Hevea accessions comprising different species. The results show unexpected levels of gene transfer between wild and cultivated types, implying insufficient reproductive isolation. This suggests that there is a possibility to introduce genetic variation into rubber farming, potentially increasing plant resilience and latex yield while maintaining quality. The pan-genome dataset and transcriptome analysis from this work also shed light on the genetic basis of latex production and defensive characteristics in rubber trees. Tapping, a mechanical wounding treatment that imitates the damage caused by insects burrowing into the bark, not only enhances the production of latex but also triggers the activation of genes related to defence mechanisms. Latex output increased with tapping frequency and was associated with increased expression of REF1 and SRPP1, two genes that are expressed in laticifers (latex producing cells) and highly duplicated in latex-producing plants. These findings are crucial for enhancing rubber breeding techniques and satisfying the growing global demand for natural rubber under changing environmental conditions. (Summary by Tuyelee Das, @das_tuyelee) Nature Comms 10.1038/s41467-024-51031-3
Natural rubber, primarily derived from Hevea brasiliensis, is an essential global resource, but its production threatened by environmental changes and pest pressures. Fang et al. examined genome assemblies from eight high-quality Hevea accessions comprising different species. The results show unexpected levels of gene transfer between wild and cultivated types, implying insufficient reproductive isolation. This suggests that there is a possibility to introduce genetic variation into rubber farming, potentially increasing plant resilience and latex yield while maintaining quality. The pan-genome dataset and transcriptome analysis from this work also shed light on the genetic basis of latex production and defensive characteristics in rubber trees. Tapping, a mechanical wounding treatment that imitates the damage caused by insects burrowing into the bark, not only enhances the production of latex but also triggers the activation of genes related to defence mechanisms. Latex output increased with tapping frequency and was associated with increased expression of REF1 and SRPP1, two genes that are expressed in laticifers (latex producing cells) and highly duplicated in latex-producing plants. These findings are crucial for enhancing rubber breeding techniques and satisfying the growing global demand for natural rubber under changing environmental conditions. (Summary by Tuyelee Das, @das_tuyelee) Nature Comms 10.1038/s41467-024-51031-3
Keystone metabolites influence rhizosphere metabolomes and microbiomes
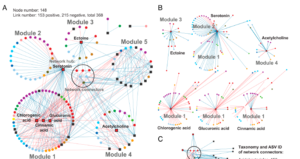 Rhizosphere interactions between plants and microbes are essential for nitrogen cycling, stress tolerance, and plant health in general. Metabolites secreted by plant roots can greatly influence microbial community composition, although how different environmental conditions impact these interactions is not fully understood. Baker et al. investigated how rhizosphere metabolites affect microbial groups in switchgrass (Panicum virgatum) when the plant is exposed to different amounts of water and nutrients. Although phosphorus and water reduction had minimal impact on microbial diversity, the metabolite profiles changed significantly. Aromatic acids like chlorogenic and caffeic acids were abundant in nitrogen-limited soils and were linked to several microbial groups. Additionally, serotonin was identified as a key metabolite that affects both plant development and microbial composition, specifically inhibiting bacteria while enhancing root length and biomass. Acetylcholine and ectoine, previously unstudied in soil, also affected microbial moisture stress responses. The results show that “keystone metabolites” like serotonin and chlorogenic acid enhance resistance to abiotic stress and influence microbial community formation in the bioenergy crop switchgrass. (Summary by Tuyelee Das, @das_tuyelee) PNAS 10.1073/pnas.2303439121
Rhizosphere interactions between plants and microbes are essential for nitrogen cycling, stress tolerance, and plant health in general. Metabolites secreted by plant roots can greatly influence microbial community composition, although how different environmental conditions impact these interactions is not fully understood. Baker et al. investigated how rhizosphere metabolites affect microbial groups in switchgrass (Panicum virgatum) when the plant is exposed to different amounts of water and nutrients. Although phosphorus and water reduction had minimal impact on microbial diversity, the metabolite profiles changed significantly. Aromatic acids like chlorogenic and caffeic acids were abundant in nitrogen-limited soils and were linked to several microbial groups. Additionally, serotonin was identified as a key metabolite that affects both plant development and microbial composition, specifically inhibiting bacteria while enhancing root length and biomass. Acetylcholine and ectoine, previously unstudied in soil, also affected microbial moisture stress responses. The results show that “keystone metabolites” like serotonin and chlorogenic acid enhance resistance to abiotic stress and influence microbial community formation in the bioenergy crop switchgrass. (Summary by Tuyelee Das, @das_tuyelee) PNAS 10.1073/pnas.2303439121
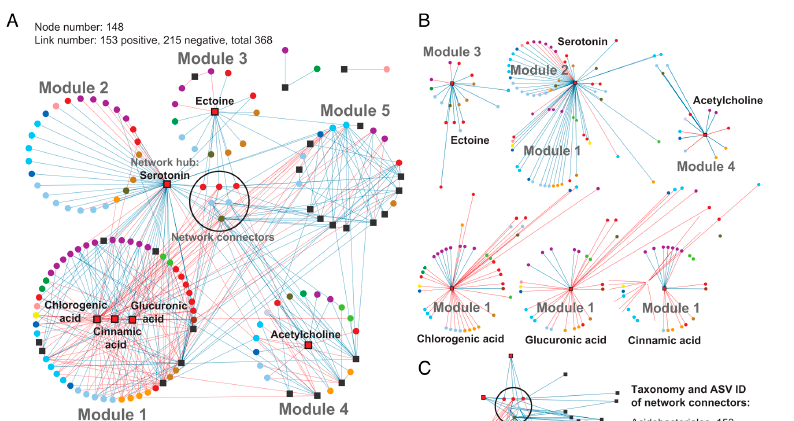 Rhizosphere interactions between plants and microbes are essential for nitrogen cycling, stress tolerance, and plant health in general. Metabolites secreted by plant roots can greatly influence microbial community composition, although how different environmental conditions impact these interactions is not fully understood. Baker et al. investigated how rhizosphere metabolites affect microbial groups in switchgrass (Panicum virgatum) when the plant is exposed to different amounts of water and nutrients. Although phosphorus and water reduction had minimal impact on microbial diversity, the metabolite profiles changed significantly. Aromatic acids like chlorogenic and caffeic acids were abundant in nitrogen-limited soils and were linked to several microbial groups. Additionally, serotonin was identified as a key metabolite that affects both plant development and microbial composition, specifically inhibiting bacteria while enhancing root length and biomass. Acetylcholine and ectoine, previously unstudied in soil, also affected microbial moisture stress responses. The results show that “keystone metabolites” like serotonin and chlorogenic acid enhance resistance to abiotic stress and influence microbial community formation in the bioenergy crop switchgrass. (Summary by Tuyelee Das, @das_tuyelee) PNAS 10.1073/pnas.2303439121
Rhizosphere interactions between plants and microbes are essential for nitrogen cycling, stress tolerance, and plant health in general. Metabolites secreted by plant roots can greatly influence microbial community composition, although how different environmental conditions impact these interactions is not fully understood. Baker et al. investigated how rhizosphere metabolites affect microbial groups in switchgrass (Panicum virgatum) when the plant is exposed to different amounts of water and nutrients. Although phosphorus and water reduction had minimal impact on microbial diversity, the metabolite profiles changed significantly. Aromatic acids like chlorogenic and caffeic acids were abundant in nitrogen-limited soils and were linked to several microbial groups. Additionally, serotonin was identified as a key metabolite that affects both plant development and microbial composition, specifically inhibiting bacteria while enhancing root length and biomass. Acetylcholine and ectoine, previously unstudied in soil, also affected microbial moisture stress responses. The results show that “keystone metabolites” like serotonin and chlorogenic acid enhance resistance to abiotic stress and influence microbial community formation in the bioenergy crop switchgrass. (Summary by Tuyelee Das, @das_tuyelee) PNAS 10.1073/pnas.2303439121