NLR-Annotator: a tool for de novo annotation of intracellular immune receptor repertoire
Wei Zhang
ORCID: 0000-0002-5092-643X
Department of Plant Pathology, Kansas State University, 1712 Claflin Road, Throckmorton Hall, Manhattan, KS, 66506, USA
Nucleotide-binding domain leucine-rich repeat (NLR) proteins serve as intracellular immune receptors in plants to recognize different types of effectors delivered by pathogens from all kingdoms. NLR genes form the largest plant disease resistance (R) gene family. NLR proteins share conserved NB-ARC domains at the N-terminus and variable leucine-rich repeat domains at the C-terminus for effector targeting specificity (Monteiro and Nishimura, 2018). During the arms race with pathogens, plant genomes have accumulated numerous NLR genes with great diversity, including both copy number variations across species, structural variation, and single nucleotide polymorphisms, especially at effector targeted C-terminal domains. The NLR gene repertoires of plants represent valuable agronomic traits for durable and broad-spectrum resistance in breeding crops such as wheat (Triticum aestivum). However, a lack of evolutionary and functional conservation together with the genetic diversity dramatically increases the difficulty in genome-wide identification and description of NLR repertoires across the plant kingdom.
Can we speed up the discovery of NLR genes by taking advantage of genomic sequencing data? Annotating NLR genes in a whole genome is the most efficient method in high-throughput identification of NLR genes. Long-read sequencing techniques enable the accurate assembly of genomic regions harboring repeated and clustered NLR genes. However, annotating NLR genes remains as painful as it was 20 years ago. Although many tools are available for automated gene annotation, they are developed based on the conserved domains/motifs with minimal manual curation. Such tools are unable to accurately identify all NLR genes because of their natural diversity, and their repeated and clustered genomic distribution. These difficulties are especially true for many economically important crops, as their genomes frequently undergo duplication during domestication. Another difficulty with these techniques is that automated gene annotation tools rely on RNA-Seq data for curation. Because NLR genes are commonly expressed primarily during infection, annotations that are based on transcriptomic data in the absence of infection will generally miss the majority of these genes.
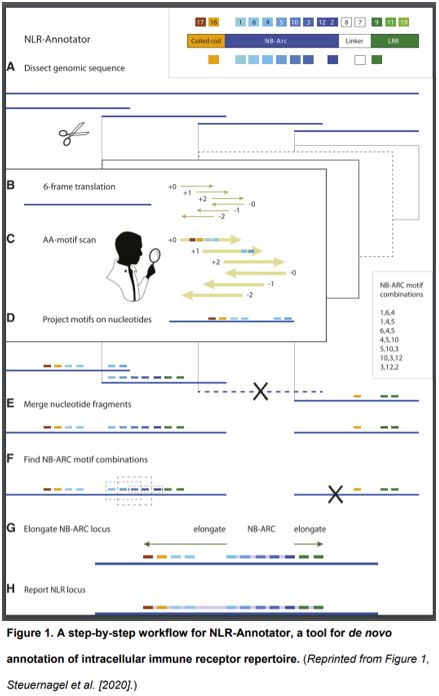 In this issue of Plant Physiology, Steuernagel et al. (2020) describe NLR-Annotator, a tool for de novo annotation of NLR genes in plant genomic data and demonstrate how it may be applied to explore the NLR repertoire in the bread wheat genome. NLR-Annotator is an update of an earlier software package (NLR-parser, Steuernagel et al., 2015) that addresses the drawbacks of relying on prior definitions of a gene model for plant NLR annotation. The new pipeline first dissects a genome into 20-kb fragments with short overlaps. Such dissected DNA fragments are further translated in all six reading frames for screening the NB-ARC associated motifs. After merging of the targeted fragments, the NB-ARC motifs are combined and used as a seed to search the DNA sequences upstream and downstream for additional NLR-associated motifs, such as coiled-coil domains or leucine-rich repeat domains. By combining all reported NLR locus, NLR-Annotator can thereby annotate the NLR repertoire for a whole genome (https://github.com/steuernb/NLR-Annotator, Figure 1).
In this issue of Plant Physiology, Steuernagel et al. (2020) describe NLR-Annotator, a tool for de novo annotation of NLR genes in plant genomic data and demonstrate how it may be applied to explore the NLR repertoire in the bread wheat genome. NLR-Annotator is an update of an earlier software package (NLR-parser, Steuernagel et al., 2015) that addresses the drawbacks of relying on prior definitions of a gene model for plant NLR annotation. The new pipeline first dissects a genome into 20-kb fragments with short overlaps. Such dissected DNA fragments are further translated in all six reading frames for screening the NB-ARC associated motifs. After merging of the targeted fragments, the NB-ARC motifs are combined and used as a seed to search the DNA sequences upstream and downstream for additional NLR-associated motifs, such as coiled-coil domains or leucine-rich repeat domains. By combining all reported NLR locus, NLR-Annotator can thereby annotate the NLR repertoire for a whole genome (https://github.com/steuernb/NLR-Annotator, Figure 1).
The quality of any annotation is evaluated by two parameters: sensitivity and specificity. The sensitivity of an annotation in this case is determined as the ratio of identified NLR genes to all NLR genes. The specificity of the annotation is the ratio of correctly identified NLRs to all identified NLRs. The NLR-Annotator exhibits both high sensitivity and specificity when applied to the Arabidopsis (Arabidopsis thaliana) genome, which is usually used as a gold standard because of its well-characterized NLR genes (Meyers et al., 2003; Van de Weyer et al., 2019). Comparative analysis of NLR repertoires across crop cultivars and other plant species requires the universal application of NLR gene annotation tools. Steuernagel et al. (2020) successfully applied NLR-Annotator to eight economic-important crop genomes, including one food and industrial resource crop, soybean (Glycine max), two cereal-related crops, maize (Zea mays) and purple false brome (Brachypodium distachyon), and five horticultural crops, including cucumber (Cucumis sativus) and potato (Solanum tuberosum). These demonstrate the broad applicability of NLR-Annotator across diverse plant taxa in phylogenetic construction using NLR genes identified with the same standard.
With NLR-Annotator in hand, the group next explored the NLR repertoire in Triticum aestivum by annotating NLR genes in a Chinese Spring cultivar with a high-quality genome assembly (Consortium IWGSC, 2018). They identified 3,400 NLR loci and 1,560 complete NLRs, and they found some intriguing features of the NLR repertoire in the bread wheat. For example, NLR loci proved to distribute predominantly across all chromosomes at their telomere regions, and half of them cluster together. The genomic arrangement pattern likely links with the evolutionary mechanisms underlying NLR gene expansion within a species. There are around 8% of proteins across the whole genome with integrated domains that encode proteins acting as a decoy or bait. The decoy proteins specialize solely in perception of the effector by the NLR protein by mimicking the effector targets and they are novel candidate genes for crop resistance breeding (van der Hoorn and Kamoun, 2008). The analysis in wheat also revealed the sequences of the NLR genes, their functional and evolutionary relationships. Finally, the wheat NLR repertoire allowed the exploration of whole-genome expression profiles of NLR genes, which showed a majority of NLRs with low expression and stress induction features.
In brief, with the NLR-Annotator pipeline of Steuernagel et al. (2020), researchers have a tool for the rigorous and reproducible annotation of NLRs across plant taxonomic clades. The species-level identification and description of NLR repertoires enable the phylogenetic construction of NLR gene families, which will provide insights on functional and evolutionary relationships among NLR genes. This knowledge will be essential for future advances that harness NLRs in economically important crops through breeding and genome editing.
References
Consortium IWGSC (2018) Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361
Meyers BC, Kozik A, Griego A, Kuang H, Michelmore RW (2003) Genome-wide analysis of NBS-LRR-encoding genes in Arabidopsis. Plant Cell 15: 809-834
Monteiro F, Nishimura MT (2018) Structural, Functional, and Genomic Diversity of Plant NLR Proteins: An Evolved Resource for Rational Engineering of Plant Immunity. Annu Rev Phytopathol 56: 243-267
Steuernagel B, Jupe F, Witek K, Jones JD, Wulff BB (2015) NLR-parser: rapid annotation of plant NLR complements. Bioinformatics 31: 1665-1667
Steuernagel B, Witek K, Krattinger SG, Ramirez-Gonzalez RH, Schoonbeek HJ, Yu G, Baggs E, Witek A, Yadav I, Krasileva KV, Jones JD, Uauy C, Keller B, Ridout CJ, Wulff BB (2020) The NLR-Annotator tool enables annotation of the intracellular immune receptor repertoire. Plant Physiol https://doi.org/10.1104/pp.19.01273
Van de Weyer AL, Monteiro F, Furzer OJ, Nishimura MT, Cevik V, Witek K, Jones JDG, Dangl JL, Weigel D, Bemm F (2019) A Species-Wide Inventory of NLR Genes and Alleles in Arabidopsis thaliana. Cell 178: 1260-1272.e1214
van der Hoorn RA, Kamoun S (2008) From Guard to Decoy: a new model for perception of plant pathogen effectors. Plant Cell 20: 2009-2017