Dynamic N1-methyladenosine in plant messenger RNA
Wei Zhang
ORCID: 0000-0002-5092-643X
Department of Plant Pathology, Kansas State University, 1712 Claflin Road, Throckmorton Hall, Manhattan, KS, 66506, USA
Deposition of different chemical groups onto RNA generates diverse RNA modifications. All types of prokaryotic and eukaryotic RNA can undergo modifications. For example, methylation of the adenosine bases at the nitrogen-6 or nitrogen-1 position generates N6-methyladenosine (m6A) and N1-methyladenosine (m1A), which are common in transfer RNAs (tRNAs) and ribosome RNAs (rRNAs), and were recently identified in messenger RNAs (mRNAs) (Roundtree et al., 2017). In contrast to the epigenetic regulation of gene expression through chemical modification of DNA and histone proteins, RNA modifications have only recently been shown to have the potential to affect most post-transcriptional steps in gene expression. Such functionally relevant changes constitute the epitranscriptome. mRNA modifications change the mRNA structure, which affects its interaction with RNA-dependent proteins during nuclear export and degradation. Modifications in mRNA and/or tRNA affect codon–anticodon interactions and translation efficiency. The well-established m6A modification of mRNA exhibits broad effects on mRNA localization, stability, translation, and alternative splicing in yeast, mammals, and plants during development and in challenging environments (Zhao et al., 2017). Only recently was m1A revealed in mammalian mRNAs with complementary effects on gene regulation (Dominissini et al., 2016; Li et al., 2016). Whether m1A is commonly present in plant mRNAs and its possible functions are poorly understood.
Discovering m6A and m1A prevalent in mRNA benefits from the development of methods that can specifically and sensitively detect and quantify mRNA modifications. Quantitative mass spectrometry (LC-MS/MS) enables direct analysis of the ratio of methylated to non-methylated adenosine in purified mRNA. Immunoblotting techniques, such as dot blotting, allow indirect detection of mRNA modifications by immunoprecipitation of antibodies that specifically bind to target RNA modifications. Immunoprecipitation coupled with high-throughput sequencing exhibits a great potential to detect RNA modifications at the whole-transcriptome level (Meyer and Jaffrey, 2014). Such epitranscriptomic data depict distribution patterns and positional preferences of mRNA modifications, providing insight into their functions in the regulation of gene expression.
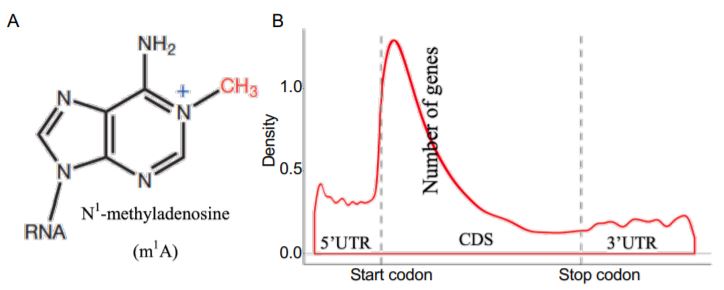
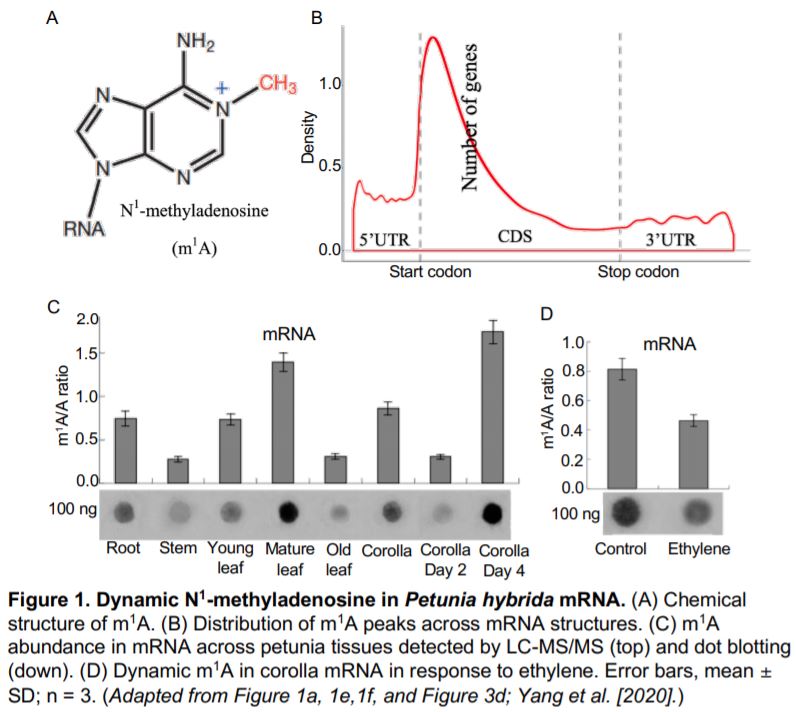 In this issue of Plant Physiology, (Yang et al., 2020) combined LC-MS/MS, dot blotting, and methylated RNA immunoprecipitation sequencing (MeRIP-seq or m1A-seq) to explore the dynamics of m1A and associated pathways in the model plant Petunia hybrida. To determine whether m1A (Figure 1A) is present in plants, Yang et al. conducted dot blotting and LC-MS/MS on total RNA, rRNA, and mRNA purified from petunia. They detected higher levels of m1A modification in the mRNA than in the rRNA. By m1A-seq, they identified 4,993 m1A peaks from 3,231 expressed genes, which covered 13.6% of 23,769 expressed genes in the petunia corolla. Interestingly, m1A peaks preferentially associated with several motifs near the start codon and within coding sequences (CDSs) when compared with other mRNA structures, such as the 5¢ untranslated region (UTR), 3¢UTR, and the stop codon (Figure 1B). This m1A positional bias in mRNA is similar to the 5-methylcytosine (m5C) modification in Arabidopsis thaliana but different from the m6A modification, which is preferentially found in the stop codon in Arabidopsis and human cells. The distinct distribution pattern among mRNA modifications may be linked to their unique functions in gene expression.
In this issue of Plant Physiology, (Yang et al., 2020) combined LC-MS/MS, dot blotting, and methylated RNA immunoprecipitation sequencing (MeRIP-seq or m1A-seq) to explore the dynamics of m1A and associated pathways in the model plant Petunia hybrida. To determine whether m1A (Figure 1A) is present in plants, Yang et al. conducted dot blotting and LC-MS/MS on total RNA, rRNA, and mRNA purified from petunia. They detected higher levels of m1A modification in the mRNA than in the rRNA. By m1A-seq, they identified 4,993 m1A peaks from 3,231 expressed genes, which covered 13.6% of 23,769 expressed genes in the petunia corolla. Interestingly, m1A peaks preferentially associated with several motifs near the start codon and within coding sequences (CDSs) when compared with other mRNA structures, such as the 5¢ untranslated region (UTR), 3¢UTR, and the stop codon (Figure 1B). This m1A positional bias in mRNA is similar to the 5-methylcytosine (m5C) modification in Arabidopsis thaliana but different from the m6A modification, which is preferentially found in the stop codon in Arabidopsis and human cells. The distinct distribution pattern among mRNA modifications may be linked to their unique functions in gene expression.
The m1A modification in mRNA showed a spatio-temporal distribution in petunia. Dot blotting and LC-MS/MS revealed that the m1A abundance in mRNA differed among roots, stems, leaves, and corolla (Figure 1C). m1A levels also depend on developmental stage, with highest levels in mature leaves and senescent corollas. Although m1A levels are positively correlated with protein production in mammals (Li et al., 2016), the m1A methylome and transcriptome did not show any linear correlation in the plant corolla. Since m1A levels are higher in senescent tissues, Yang et al. hypothesized that the m1A modification may function in senescence. They treated corollas with ethylene, which accelerates senescence, and observed reduced m1A levels in corolla mRNA by dot blotting and LC-MS/MS (Figure 1D). m1A-seq analysis revealed that ethylene treatment caused a whole-genome redistribution of m1A in the mRNA pool. Some mRNAs showed qualitative variation with either loss or gain of m1A modifications, while others showed quantitative variation with either increased or reduced m1A levels. Interestingly, the changes in the m1A methylome and transcriptome resulting from the ethylene treatment showed a slightly but significantly positive correlation, indicating a link between m1A modification and gene expression. Thus, plants can modulate m1A levels in mRNA to coordinate tissue- or developmental stage-specific signals and stimuli from the environment.
The dynamics of m1A in mRNA are precisely balanced by the action of writers (methyltransferases) and erasers (demethylases) (Zhao et al., 2017). In yeast and mammals, tRNA methyltransferase transfers methyl groups from S-adenosylmethionine to specific RNA positions. Yang et al. identified a tRNA-specific methyltransferase 61A (PhTRMT61A) with in vitro methyltransferase capacity. PhTRMT61A is transcribed in leaves, roots, stems, and corollas, and can be repressed by ethylene. The encoded protein localizes to the nucleus, suggesting its function in the methylation of nuclear RNA. Loss of PhTRMT61A caused a chlorotic and wrinkled leaf phenotype with decreased leaf chlorophyll content and small cells. Thus, PhTRMT61A functions in cellular processes by regulating m1A levels in petunia nuclear RNA.
Taken together, Yang et al. (2020) revealed the dynamics of m1A modification in petunia mRNAs in diverse tissues and developmental stages. Dynamic m1A profiling is finely tuned by the writer PhTRMT61A and the eraser, although the identity of the latter remains unclear in petunia. These findings provide insight into the complex transcriptional, epigenetic, and epitranscriptomic mechanisms controlling gene transcription and translation in response to signals from within the plant and from the external environment.
References
Dominissini D, Nachtergaele S, Moshitch-Moshkovitz S, Peer E, Kol N, Ben-Haim MS, Dai Q, Di Segni A, Salmon-Divon M, Clark WC, Zheng G, Pan T, Solomon O, Eyal E, Hershkovitz V, Han D, Doré LC, Amariglio N, Rechavi G, He C (2016) The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature 530: 441-446
Li X, Xiong X, Wang K, Wang L, Shu X, Ma S, Yi C (2016) Transcriptome-wide mapping reveals reversible and dynamic N(1)-methyladenosine methylome. Nat Chem Biol 12: 311-316
Meyer KD, Jaffrey SR (2014) The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat Rev Mol Cell Biol 15: 313-326
Roundtree IA, Evans ME, Pan T, He C (2017) Dynamic RNA Modifications in Gene Expression Regulation. Cell 169: 1187-1200
Zhao BS, Roundtree IA, He C (2017) Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol 18: 31-42
")