A novel way to conduct genome wide association studies for secondary metabolites
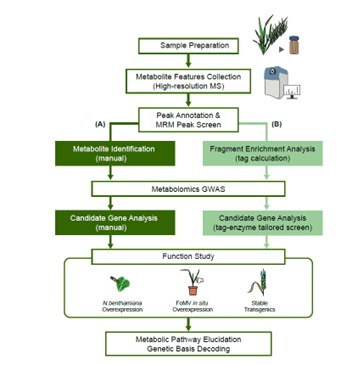
Plants produce many specialized metabolites that are often associated with increased plant fitness. Metabolic genome wide association studies (mGWAS) identify enzymes involved in specialized metabolite biosynthesis. However, this relies on identifying metabolites from mass spectrometry data which is often time and labor intensive. Here, Zhu et al. bypassed this step by identifying structural modifications rather than the entire metabolite. They used liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS) on leaf extracts from 391 diverse wheat accessions and identified six modifications in the metabolites: hydration, glycosylation, methylation, acetylation, decarboxylation and acylation. This information was fed into a mGWAS pipeline that identified 3594 loci associated with the modifications. To reduce this number, they excluded loci that did not contain an enzyme capable of performing the modification and loci that did not have single nucleotide polymorphisms (SNPs) in the coding sequence or promoter of this gene. This successfully reduced the 3594 loci to 499 candidates. The authors showed that the function of these candidates can be validated in wheat using a Foxtail mosaic virus transient expression system. This strategy is a vast improvement on the traditional mGWAS method as it removes the time-consuming step of metabolite identification and is an unbiased way of identifying novel gene candidates. (Summary by Rose McNelly @Rose_McN) Plant Cell 10.1093/plcell/koad286