Mutagenomics: The Future of Genetic Screens
Ananya Mukherjee
ORCID ID: 0000-0003-1802-1806
University of Nebraska Lincoln
Genetic screens are a remarkable way of identifying mutations affecting various aspects of plant growth and development. Arabidopsis (Arabidopsis thaliana) (T)-DNA insertion lines enable rapid identification of mutant genes but are tedious to create in a mutant background of choice. Chemical mutagens such as ethyl methanesulfonate (EMS) create hundreds of allelic variants in any background of choice and potentially avoid lethality of null alleles. However, cloning these variants can be challenging. Additionally, EMS mutagenesis causes many single-nucleotide polymorphisms (SNPs) that are not causative for the mutation of interest. Backcrossing allows the identification of causative mutations but is labor intensive. High-throughput sequencing has facilitated the development of several pipelines that involve cloning genes corresponding to allelic variants, thereby reducing the need for extensive backcrossing (Michelmore et al., 1991; Cluis et al., 2004; Austin et al., 2011; Nordström et al., 2013). Although powerful, only a limited number of lines are generally screened by these methods. For many pathways of interest, prior genetic screens have already identified multiple genes. The use of enhancer/suppressor screens combined with high-throughput analysis of identified mutations has the potential to identify novel elements in a pathway of interest.
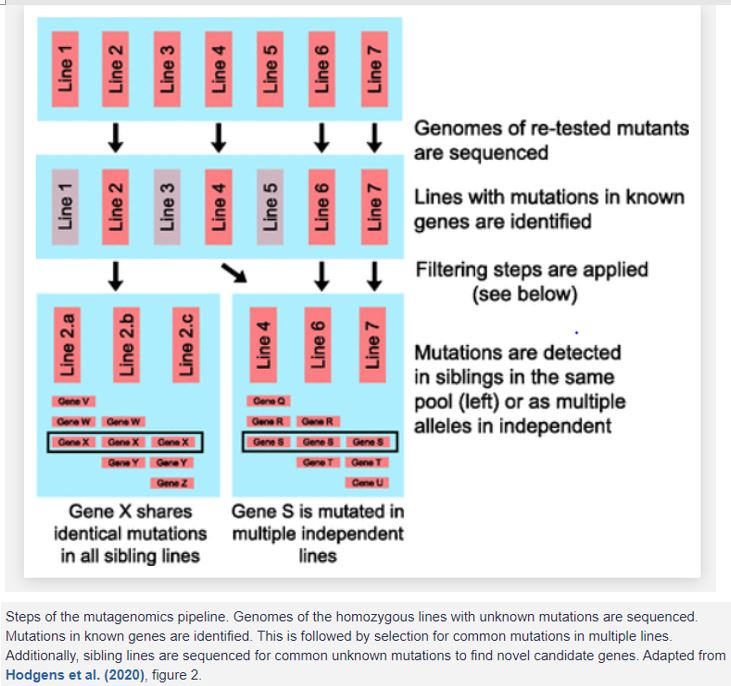 In this issues of Plant Physiology, Hodgens et al. (2020) describe a strategy they developed and named mutagenomics. This streamlined process aims to find casual mutations that can help identify newer candidate genes in a pathway. Mutagenomics can be useful in both well-established pathways with several known components and in newer processes with few known components.
In this issues of Plant Physiology, Hodgens et al. (2020) describe a strategy they developed and named mutagenomics. This streamlined process aims to find casual mutations that can help identify newer candidate genes in a pathway. Mutagenomics can be useful in both well-established pathways with several known components and in newer processes with few known components.
The authors have used the well-established cytokinin pathway as an example for the screen and used a background that is sensitized to small perturbations to cytokinin. To screen for unknown components of the cytokinin pathway in Arabidopsis, they started with a strain that is genetically sensitized to modest changes in cytokinin responsiveness. This loss-of-function double mutant for Histidine phosphotransfer protein2 and –3 was then EMS mutagenized and screened for root length. A root elongation assay using 0.1 µm 6-benzylaminopurine, a synthetic cytokinin, indicated decreased cytokinin sensitivity based on root length. They identified 272 putative mutants from 78 individual pools. They then used their mutagenomics pipeline to evaluate the putative mutants (Fig. 1).
Several known cytokinin-responsive genes were identified, showing that the screening strategy was successful. For instance, 13 alleles (eight missense, four nonsense, and one for a splice site) were identified for His kinase (ahk4), which is a predominant cytokinin receptor for root response (Riefler et al., 2006). However, since the goal was to identify additional candidate genes for the cytokinin pathway, the known genes identified in the screen were eliminated. Homozygous lines with altered cytokinin sensitivity were first sequenced. This was followed by selecting protein-altering SNPs (PA-SNPs) that were homozygous.
The next step was to identify mutations that had occurred in multiple lines. The idea was to filter multiple putative mutants that had occurred at a frequency above that expected by chance. Simulation modeling was performed to determine the probability of independent mutations in these lines. It was important to note that the number of times a gene can be mutated depends on its size and number of lines screened. Genes were thus analyzed based on quartiles of gene sizes, with the smallest quartile having a gene size less than 590 coding bases. This filtering step allowed the authors to identify and focus on PA-SNPs overrepresented in the population, in genes that were most likely responsible for cytokinin hyposensitivity.
The third step of the pipeline is what makes mutagenomics unique. Up to this point, all the identified mutations had been in known genes. Next, the authors analyzed 22 lines where a causative gene had not been identified. To screen for novel candidates, additional lines (siblings) from the same pools were sequenced. The principle behind this step was to narrow the number of potential candidate genes by considering only the common PA-SNPs among siblings. The idea was that the noncausative PA-SNPs will segregate randomly, whereas those associated with the phenotype will be present in the additional sequences. For instance, if a line with 285 PA-SNPs is selected, sequencing four siblings in theory reduces the number of overlapping PA-SNPs to five or fewer. A line with 13 PA-SNPs was selected for sibling sequencing. Of the six PA-SNPs found in the siblings, a PA-SNP in ELONGATED HYPOCOTYL5 (HY5) was selected for further analysis. HY5 is a transcription factor that has been suggested to play a role in cytokinin sensitivity (Cluis et al., 2004). Further characterization showed strong overlap among the binding sites of HY5 and other known cytokinin transcription factors, demonstrating the power of the mutagenomics pipeline to provide new insight into well-studied pathways. Further analysis of the remaining lines could potentially add more to what we already know about this pathway.
In conclusion, mutagenomics has the advantage of rapidly parallel processing several mutant lines and identifying novel genes that play roles in a biological pathway. The application of this pipeline is not limited to Arabidopsis and can be easily extended to other model and nonmodel organisms.
REFERENCES
Austin RS, Vidaurre D, Stamatiou G, Breit R, Provart NJ, Bonetta D, Zhang J, Fung P, Gong Y, Wang PW (2011) Next‐generation mapping of Arabidopsis genes. The Plant Journal 67: 715-725
Cluis CP, Mouchel CF, Hardtke CS (2004) The Arabidopsis transcription factor HY5 integrates light and hormone signaling pathways. Plant J 38: 332-347
Hodgens C, Chang N, Schaller GE, Kieber JJ (2020) Mutagenomics: A rapid, high-throughput method to identify causative mutations from a genetic screen. Plant Physiology: pp.00609.02020 https://doi.org/10.1104/pp.20.00609
Michelmore RW, Paran I, Kesseli RV (1991) Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proceedings of the National Academy of Sciences of the United States of America 88: 9828-9832
Nordström KJ, Albani MC, James GV, Gutjahr C, Hartwig B, Turck F, Paszkowski U, Coupland G, Schneeberger K (2013) Mutation identification by direct comparison of whole-genome sequencing data from mutant and wild-type individuals using k-mers. Nature biotechnology 31: 325-330
Riefler M, Novak O, Strnad M, Schmülling T (2006) Arabidopsis cytokinin receptor mutants reveal functions in shoot growth, leaf senescence, seed size, germination, root development, and cytokinin metabolism. The Plant cell 18: 40-54

