When binding isn’t enough: Predicting TF activity through genomic neighborhoods
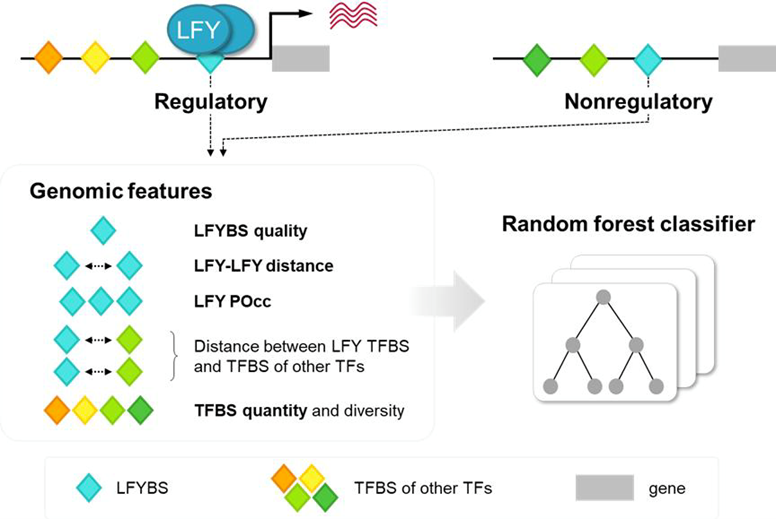 Transcriptional regulation is fundamental for coordinating cellular activities in response to developmental programs and environmental cues. Certain transcription factors (TFs) control the expression of hundreds of genes and are thus considered especially important — these are referred to as master regulators. However, predicting the regulatory activity of a TF remains challenging. While techniques such as ChIP-seq and DAP-seq are routinely used to identify TF binding sites, many of these sites may be non-regulatory. Temporal and tissue-specific expression adds further complexity to sampling and interpretation. Turchi and colleagues addressed this issue by adopting a new machine learning model that uses “genomic context” as input. This model incorporates not only the predicted TF binding sites and their spacing but also — crucially — the presence of other TFs that may influence the activity of the target TF. Using LEAFY (LFY) — a master regulator of flower development in Arabidopsis — as a case study, the authors constructed a dataset to evaluate the model’s ability to distinguish between regulatory and non-regulatory LFY binding sites. Their results show that incorporating genomic context features significantly improves predictive performance over traditional models that rely solely on TF occupancy or position weight matrices. This study offers new insights into how TF activity is regulated and how interactions among TFs contribute to gene expression control. (Summary by Ching Chan @ntnuchanlab) bioRxiv 10.1101/2025.05.23.655699
Transcriptional regulation is fundamental for coordinating cellular activities in response to developmental programs and environmental cues. Certain transcription factors (TFs) control the expression of hundreds of genes and are thus considered especially important — these are referred to as master regulators. However, predicting the regulatory activity of a TF remains challenging. While techniques such as ChIP-seq and DAP-seq are routinely used to identify TF binding sites, many of these sites may be non-regulatory. Temporal and tissue-specific expression adds further complexity to sampling and interpretation. Turchi and colleagues addressed this issue by adopting a new machine learning model that uses “genomic context” as input. This model incorporates not only the predicted TF binding sites and their spacing but also — crucially — the presence of other TFs that may influence the activity of the target TF. Using LEAFY (LFY) — a master regulator of flower development in Arabidopsis — as a case study, the authors constructed a dataset to evaluate the model’s ability to distinguish between regulatory and non-regulatory LFY binding sites. Their results show that incorporating genomic context features significantly improves predictive performance over traditional models that rely solely on TF occupancy or position weight matrices. This study offers new insights into how TF activity is regulated and how interactions among TFs contribute to gene expression control. (Summary by Ching Chan @ntnuchanlab) bioRxiv 10.1101/2025.05.23.655699