SAM-seq: A novel approach for unraveling plant epigenetic complexity
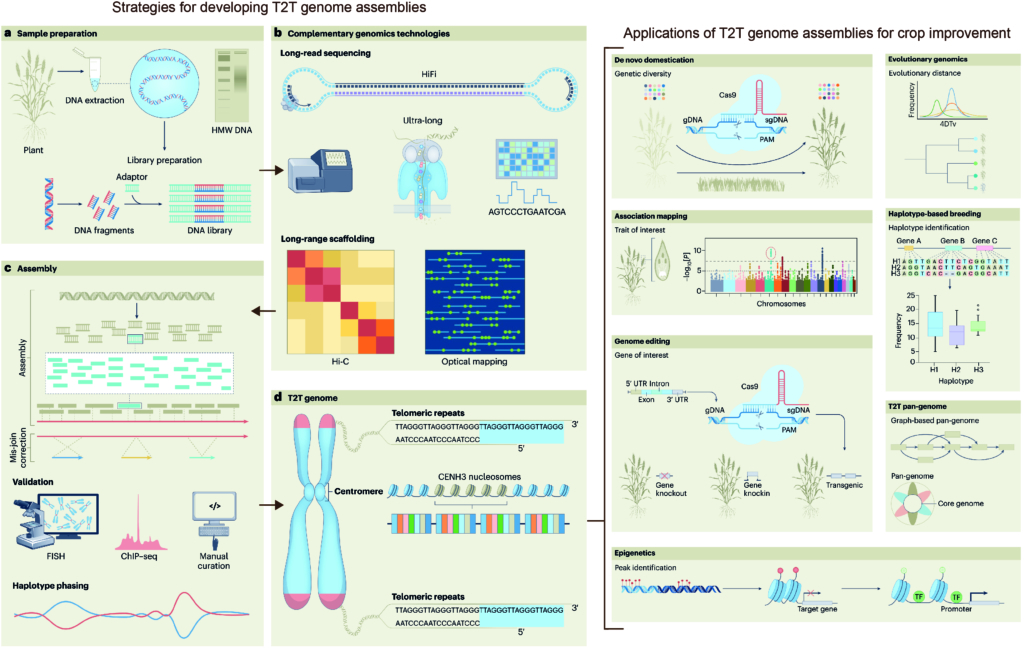
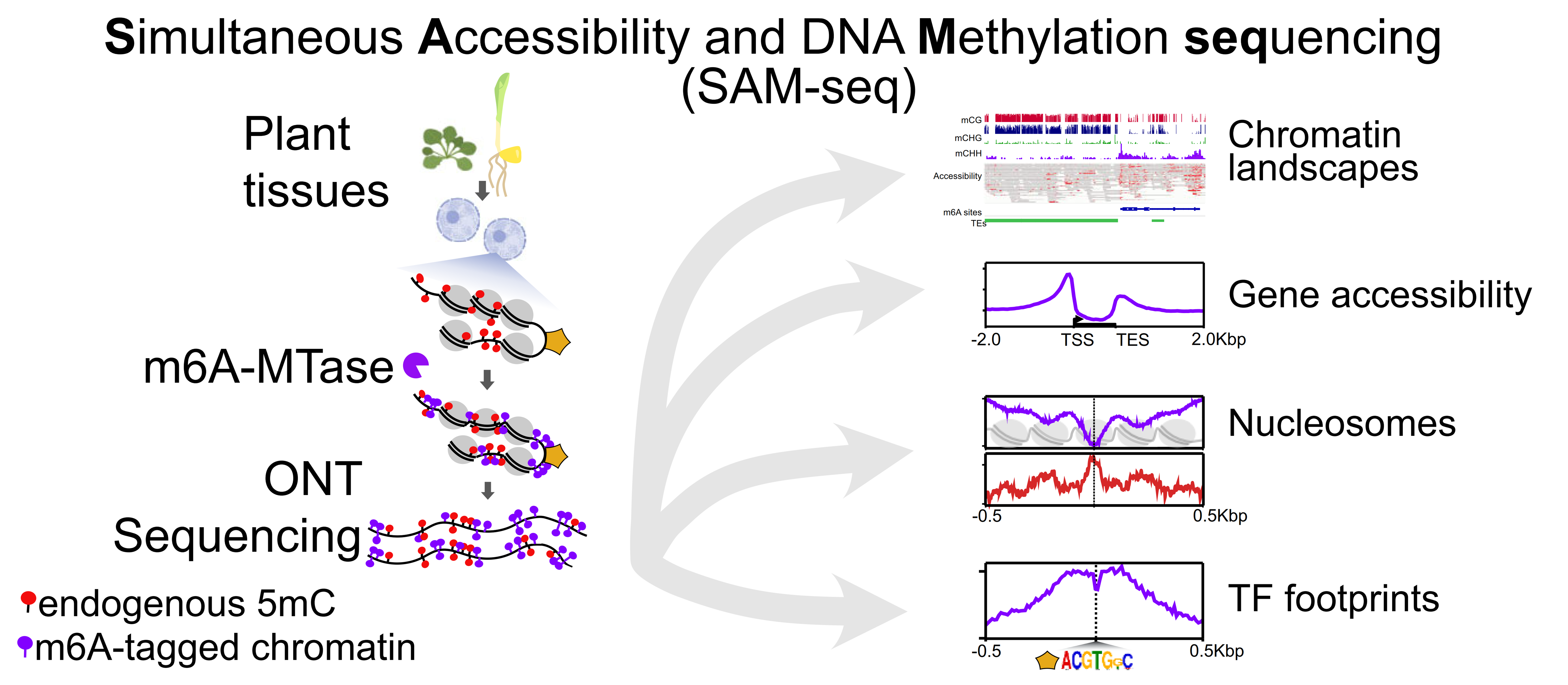 The complex interplay between nucleosome positioning, DNA methylation, and chromatin accessibility is crucial for genome regulation in eukaryotic organisms. However, current chromatin profiling methods, which rely on short-read sequencing, fail to characterize highly repetitive genomic regions and cannot detect multiple chromatin features simultaneously. In a recent study, Leduque et al. addressed these challenges by introducing a novel approach called SAM-seq (Simultaneous Accessibility and DNA Methylation sequencing). They adapted the m6A-tagged chromatin accessibility assay, SMAC-seq, for use in purified plant nuclei and employed long-read nanopore sequencing technology to assess both chromatin accessibility and DNA methylation simultaneously. SAM-seq proved effective not only in the model organism Arabidopsis but also in a complex plant genome such as maize. SAM-seq allowed for a detailed examination of accessibility and DNA methylation, particularly in subnucleosomal regions associated with genes, transposable elements, and centromeric repeats. Notably, the power and sensitivity of SAM-seq facilitated the detection of short-scale changes in accessibility and the identification of cis-regulatory regions occupied by transcription factors in vivo. Additionally, the study revealed cellular heterogeneity within chromatin domains with opposing chromatin marks, suggesting that bivalency reflects cell-specific regulatory mechanisms rather than a uniform chromatin state. SAM-seq also offers an all-in-one tool to generate de novo genome assemblies together with detailed epigenome information. Its technical simplicity, reproducibility, scalability, and cost-effectiveness make it a valuable tool for studying chromatin regulation in both model and non-model plant species. (Summary by Ileana Tossolini @IleanaDrt) Nucleic Acids Res. 10.1093/nar/gkae306
The complex interplay between nucleosome positioning, DNA methylation, and chromatin accessibility is crucial for genome regulation in eukaryotic organisms. However, current chromatin profiling methods, which rely on short-read sequencing, fail to characterize highly repetitive genomic regions and cannot detect multiple chromatin features simultaneously. In a recent study, Leduque et al. addressed these challenges by introducing a novel approach called SAM-seq (Simultaneous Accessibility and DNA Methylation sequencing). They adapted the m6A-tagged chromatin accessibility assay, SMAC-seq, for use in purified plant nuclei and employed long-read nanopore sequencing technology to assess both chromatin accessibility and DNA methylation simultaneously. SAM-seq proved effective not only in the model organism Arabidopsis but also in a complex plant genome such as maize. SAM-seq allowed for a detailed examination of accessibility and DNA methylation, particularly in subnucleosomal regions associated with genes, transposable elements, and centromeric repeats. Notably, the power and sensitivity of SAM-seq facilitated the detection of short-scale changes in accessibility and the identification of cis-regulatory regions occupied by transcription factors in vivo. Additionally, the study revealed cellular heterogeneity within chromatin domains with opposing chromatin marks, suggesting that bivalency reflects cell-specific regulatory mechanisms rather than a uniform chromatin state. SAM-seq also offers an all-in-one tool to generate de novo genome assemblies together with detailed epigenome information. Its technical simplicity, reproducibility, scalability, and cost-effectiveness make it a valuable tool for studying chromatin regulation in both model and non-model plant species. (Summary by Ileana Tossolini @IleanaDrt) Nucleic Acids Res. 10.1093/nar/gkae306