How do root hairs keep their tubular shape?
Hirano et al. uncover the secretion route for root hair shank hardening in Arabidopsis. The Plant Cell (2023).
https://doi.org/10.1093/plcell/koad240
By T. Hirano, K. Ebine, T. Ueda, T. Higaki, T. W.-Nakayama, H. Konno, H. T.-Imamura, and M. H. Sato
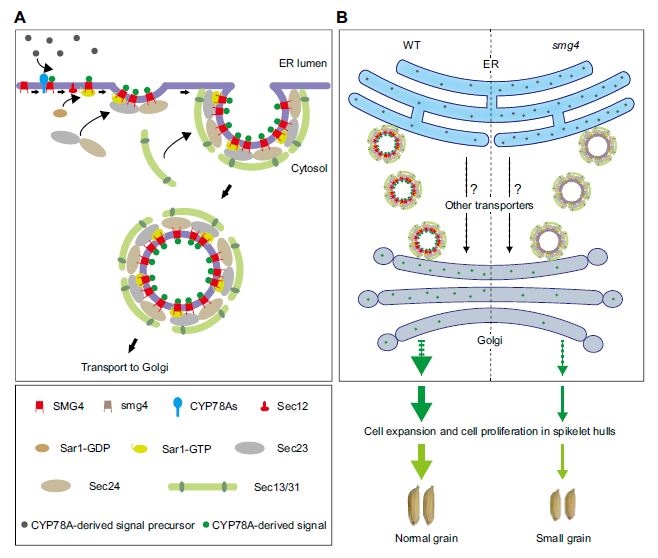
Background: Root hairs are rapidly growing tubular projections of root epidermal cells and play an essential role in plant water and nutrient uptake. Root hair formation involves tip growth and the concomitant hardening of the shank by producing a secondary cell wall layer to form an elongated tubular structure.
Question: The polarized secretion of materials in the root hair tip has been well studied. However, little is known about the secretion of secondary cell wall materials in the root hair shank.
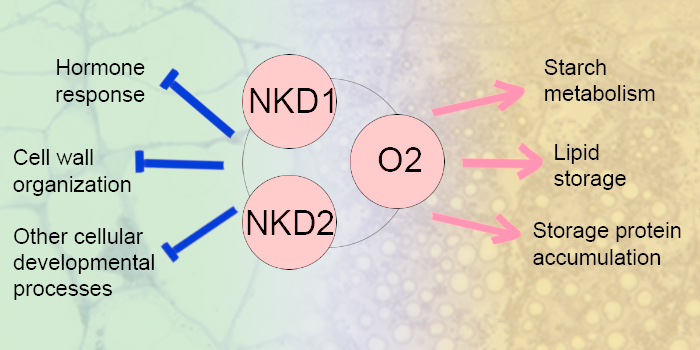
Findings: We observed increased localization of SYP123 at the plasma membrane of the subapical and shank zones compared to the tip region in elongating root hairs. Inhibition of PtdIns(3,5)P2 production impaired SYP123 localization in the plasma membrane and SYP123-mediated root hair shank hardening, and root hair elongation in syp123 mutant is insensitive to the PtdIns(3,5)P2 synthesis inhibitor. SYP123 interacts with both VAMP721 and VAMP727. The syp123 and vamp727 mutants exhibited reduced shank cell wall stiffness due to impaired secondary cell wall components deposition. In conclusion, the SYP123/VAMP727-dependent secretion system delivers secondary cell wall components for hardening the subapical zone and shank of Arabidopsis root hairs.
Next steps: Further studies are needed to confirm the detailed molecular mechanisms underlying the multiple secretion pathways for root hair morphogenesis.
Reference:
Tomoko Hirano, Kazuo Ebine, Takashi Ueda, Takumi Higaki, Takahiro Watanabe-Nakayama, Hiroki Konno, Hisako Takigawa-Imamura, and Masa H. Sato. The SYP123-VAMP727 SNARE complex is involved in the delivery of secondary cell wall components for hardening the root hair shank in Arabidopsis https://doi.org/10.1093/plcell/koad240
 Findings: White variegation in red-skinned grapevine fruits is caused by the presence of non-functional alleles of MYBA1/A2 transcription factors that naturally control anthocyanin biosynthesis. The absence of these pigments enhances a ripening-dependent regulatory network mediated by MYB24 that promotes protection against ultraviolet and high-light intensity stress. In response, white skin sections accumulate higher levels of antioxidant monoterpenes and UV-shielding flavonols; however, these compounds only partially ameliorate the detrimental effects of excessive radiation. Genes related to carotenoid metabolism, photosynthesis and other light signaling responses are bound and directly regulated by MYB24. By conducting in silico and in vitro analyses and using field-grown grapevine plants, we demonstrate that MYB24 orchestrates different specialized metabolism pathways in berry skins in response to increased levels of radiation caused by pigment depletion.
Findings: White variegation in red-skinned grapevine fruits is caused by the presence of non-functional alleles of MYBA1/A2 transcription factors that naturally control anthocyanin biosynthesis. The absence of these pigments enhances a ripening-dependent regulatory network mediated by MYB24 that promotes protection against ultraviolet and high-light intensity stress. In response, white skin sections accumulate higher levels of antioxidant monoterpenes and UV-shielding flavonols; however, these compounds only partially ameliorate the detrimental effects of excessive radiation. Genes related to carotenoid metabolism, photosynthesis and other light signaling responses are bound and directly regulated by MYB24. By conducting in silico and in vitro analyses and using field-grown grapevine plants, we demonstrate that MYB24 orchestrates different specialized metabolism pathways in berry skins in response to increased levels of radiation caused by pigment depletion. Question: We wanted to know how many lncRNAs are present in the model plant Arabidopsis (Arabidopsis thaliana), how they differ in plants from different regions, and whether the recently reported widespread epigenetic variation in Arabidopsis underlies this difference. We used many transcriptome and epigenetic sequencing datasets to answer these questions.
Question: We wanted to know how many lncRNAs are present in the model plant Arabidopsis (Arabidopsis thaliana), how they differ in plants from different regions, and whether the recently reported widespread epigenetic variation in Arabidopsis underlies this difference. We used many transcriptome and epigenetic sequencing datasets to answer these questions.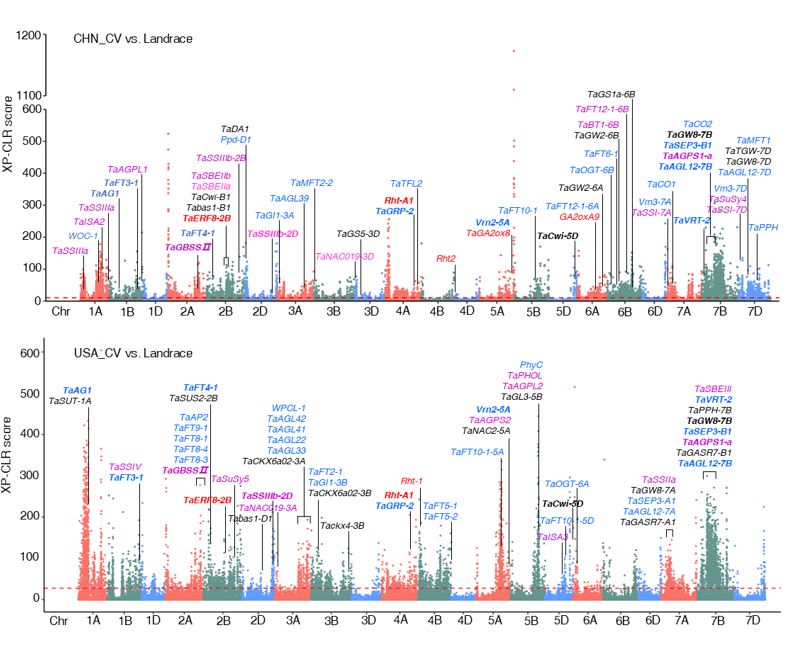 Next steps: The unique loci selected either in China or the United States can be used to develop high-performance wheat varieties in the future.
Next steps: The unique loci selected either in China or the United States can be used to develop high-performance wheat varieties in the future.