Mutation bias shapes gene evolution in Arabidopsis thaliana (bioRxiv)
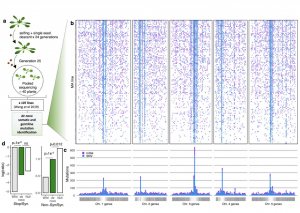 Classical evolutionary theory states that the probability of a mutation occurring is independent of fitness consequences. However, reassessment of traditional assumptions is warranted with recent discoveries showing that cytogenetic (DNA sequences and epigenetic) features can affect local mutation probabilities. Cytogenetic features have distinct states among certain gene classes which may facilitate the evolution of beneficial mutation rates. To test whether mutation rate bias can be adaptive, Monroe et al. built a high-resolution predictive model of mutation rates in Arabidopsis (see the Twitter thread here). This model was developed using 10,500 de novo mutations from sequencing 107 mutation accumulation lines alongside genome-wide data (histone modifications, chromatin accessibility, GC-content). This model resulted in three main findings. Firstly, cytogenetic features significantly predict mutation rates (e.g., lower mutation rates in GC-rich regions). Secondly, introns and untranslated regions physically distance coding sequences from mutation hotspots, with higher mutation rates in genes lacking UTRs or introns (30-40% and 90%, respectively). Finally, genes sensitive to deleterious mutations possess lower coding-region mutation rates. Intriguingly, these findings were mirrored in natural variation patterns. The authors’ explanation for these observations is that selection on mutation rate is not at the gene-level but epigenetic features differentially direct DNA repair machinery recruitment. These discoveries contradict the classical evolutionary theory that mutation probabilities are independent of gene function and fitness consequences, signifying an important breakthrough in our understanding of gene evolution. In a new approach, the authors have released a comments-enabled google doc so that the wider community can provide feedback on the manuscript, which is available here. (Summary by Caroline Dowling @CarolineD0wling) bioRxiv 10.1101/2020.06.17.156752.
Classical evolutionary theory states that the probability of a mutation occurring is independent of fitness consequences. However, reassessment of traditional assumptions is warranted with recent discoveries showing that cytogenetic (DNA sequences and epigenetic) features can affect local mutation probabilities. Cytogenetic features have distinct states among certain gene classes which may facilitate the evolution of beneficial mutation rates. To test whether mutation rate bias can be adaptive, Monroe et al. built a high-resolution predictive model of mutation rates in Arabidopsis (see the Twitter thread here). This model was developed using 10,500 de novo mutations from sequencing 107 mutation accumulation lines alongside genome-wide data (histone modifications, chromatin accessibility, GC-content). This model resulted in three main findings. Firstly, cytogenetic features significantly predict mutation rates (e.g., lower mutation rates in GC-rich regions). Secondly, introns and untranslated regions physically distance coding sequences from mutation hotspots, with higher mutation rates in genes lacking UTRs or introns (30-40% and 90%, respectively). Finally, genes sensitive to deleterious mutations possess lower coding-region mutation rates. Intriguingly, these findings were mirrored in natural variation patterns. The authors’ explanation for these observations is that selection on mutation rate is not at the gene-level but epigenetic features differentially direct DNA repair machinery recruitment. These discoveries contradict the classical evolutionary theory that mutation probabilities are independent of gene function and fitness consequences, signifying an important breakthrough in our understanding of gene evolution. In a new approach, the authors have released a comments-enabled google doc so that the wider community can provide feedback on the manuscript, which is available here. (Summary by Caroline Dowling @CarolineD0wling) bioRxiv 10.1101/2020.06.17.156752.