Techniques Becoming Obsolete in Plant Molecular Biology in 2024
The field of plant molecular biology has experienced remarkable growth in recent decades, contributed by significant milestones such as the recognition of Arabidopsis thaliana as a universal plant model, the Arabidopsis Genome Initiative in 2000 (The Arabidopsis Genome, 2000), and the 1001 Genome Initiative in 2016 (Alonso-Blanco et al., 2016). The assembly of the Arabidopsis genome, with its challenging regions like the centromeres, represented the pinnacle of genome quality at that time. Transformative techniques, including plant transformation and mutagenesis, combined with the rise of multi-omics technology, have propelled the field forward, generating extensive big data that has revolutionized traditional analysis methods.
Genetic engineering
Genetic engineering has been a cornerstone in plant biology research since the 1990s, with transfer DNA (T-DNA) sequence-indexed mutant collections playing a pivotal role in studying Arabidopsis thaliana genes (Alonso et al., 2003). Similar collections have been established in crop plants like rice, wheat, and maize. These collections provided a vast source of mutant alleles for in vivo characterization of gene function, significantly contributing to plant biology research. However, limitations on organism-specific features, transformability, and lifespan have restricted research in non-model plant species and novel target genes. The advent of targeted mutagenesis methods such as Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) (Cong et al., 2013) and Transcription Activator-like Effector Nucleases (TALENS) (Cermak et al., 2011) has revolutionized the field, allowing for the generation of custom alleles with increasing specificity and reduced off-target effects. As genome editing becomes more cost-effective, it has become a standard laboratory technique globally. Despite the continued popularity of T-DNA and Ethyl methanesulfonate (EMS)-based gene-indexed mutants, CRISPR technology has overtaken them in terms of publications since 2016 (Fig. 1A), indicating a shift towards more advanced and precise genetic manipulation methods.
Transcriptome Profiling: The Shift from Microarrays to RNA-Seq
Since the 1990s, microarrays have been pivotal in large-scale gene expression studies, providing insights into developmental processes and gene regulation. However, limitations such as background hybridization and probe design constraints have hindered their accuracy, especially for low-abundance transcripts. The advent of RNA-Seq has marked a transformative shift in transcriptome profiling (Wang et al., 2009). By directly sequencing transcripts through high-throughput technologies, RNA-Seq overcomes the limitations of microarrays, enabling the detection of novel transcripts, allele-specific expression, and splice junctions. With advancements in computational algorithms and increased affordability, RNA-Seq has become the method of choice, nearly replacing hybridization-based arrays in transcriptome profiling studies (Fig. 1B).
Comparative Proteomics: From 2DE to Mass Spectrometry-Based Methods
Two-dimensional gel electrophoresis (2DE) has been a cornerstone in proteomics studies, allowing the resolution of complex protein mixtures. This technique, popular in past decades, facilitated the large-scale analysis of protein expression patterns. Mass spectrometry-based methods have gradually supplanted 2DE, offering enhanced sophistication and performance (Aebersold and Mann, 2003). Mass spectrometry (MS) allows the identification and quantification of proteins based on their mass-to-charge ratio in a vacuum environment. This analytical technique, complemented by computational and bioinformatics tools, has simplified and made protein identification more cost-effective. The exponential growth of proteomics, predominantly fueled by MS-based methods (Fig. 1C), enables post-identification analyses, including ontology analysis, elucidating biological processes, cellular components, molecular function, post-translational modifications, and protein-protein interactions.
Perspectives
As plant molecular biology embraces these advancements, it is crucial to bid farewell to obsolete techniques. The transition from microarrays to RNA-Seq and from 2DE to mass spectrometry-based methods, coupled with the continuous evolution of genetic engineering, marks a new era characterized by increased accuracy, efficiency, and depth of analysis. Plant molecular biologists are now equipped with cutting-edge tools, including advanced genetic manipulation methods, to explore the intricacies of plant biology and unravel the mysteries encoded in the plant genome.
Figure legend
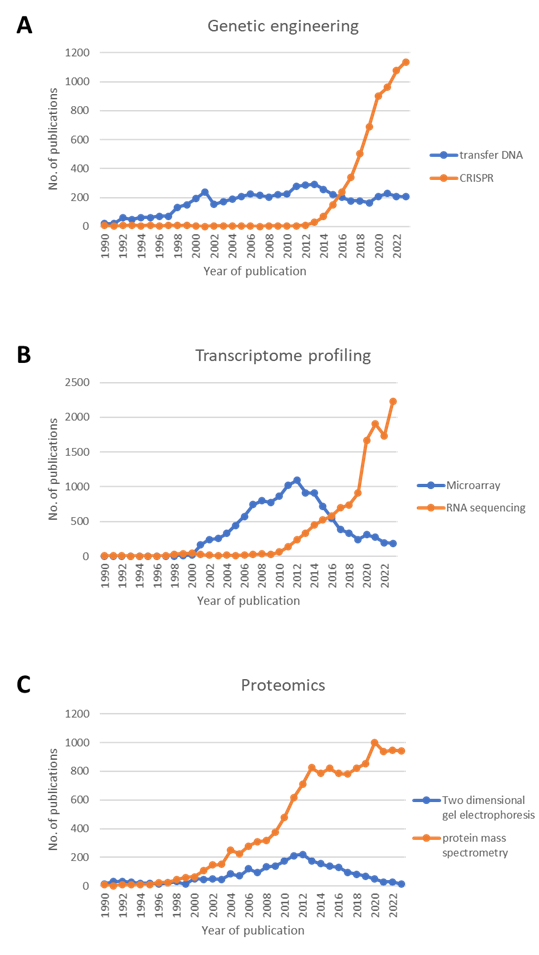
Fig. 1. Overview of Scopus-indexed publications comparing methodologies in (A) genetic engineering utilizing transfer DNA versus CRISPR; (B) transcriptome profiling employing microarray versus RNA-seq; and (C) comparative proteomics using Two-dimensional gel electrophoresis versus protein mass spectrometry.
References
- Aebersold R, Mann M. 2003. Mass spectrometry-based proteomics. Nature 422, 198-207.
- Alonso-Blanco C, Andrade J, Becker C, Bemm F, Bergelson J, Borgwardt KM, Cao J, Chae E, Dezwaan TM, Ding W, Ecker JR, Exposito-Alonso M, Farlow A, Fitz J, Gan X, Grimm DG, Hancock AM, Henz SR, Holm S, Horton M, Jarsulic M, Kerstetter RA, Korte A, Korte P, Lanz C, Lee C-R, Meng D, Michael TP, Mott R, Muliyati NW, Nägele T, Nagler M, Nizhynska V, Nordborg M, Novikova PY, Picó FX, Platzer A, Rabanal FA, Rodriguez A, Rowan BA, Salomé PA, Schmid KJ, Schmitz RJ, Seren Ü, Sperone FG, Sudkamp M, Svardal H, Tanzer MM, Todd D, Volchenboum SL, Wang C, Wang G, Wang X, Weckwerth W, Weigel D, Zhou X. 2016. 1,135 Genomes Reveal the Global Pattern of Polymorphism in <em>Arabidopsis thaliana</em>. Cell 166, 481-491.
- Alonso JM, Stepanova AN, Leisse TJ, Kim CJ, Chen H, Shinn P, Stevenson DK, Zimmerman J, Barajas P, Cheuk R, Gadrinab C, Heller C, Jeske A, Koesema E, Meyers CC, Parker H, Prednis L, Ansari Y, Choy N, Deen H, Geralt M, Hazari N, Hom E, Karnes M, Mulholland C, Ndubaku R, Schmidt I, Guzman P, Aguilar-Henonin L, Schmid M, Weigel D, Carter DE, Marchand T, Risseeuw E, Brogden D, Zeko A, Crosby WL, Berry CC, Ecker JR. 2003. Genome-Wide Insertional Mutagenesis of Arabidopsis thaliana. Science 301, 653-657.
- Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, Baller JA, Somia NV, Bogdanove AJ, Voytas DF. 2011. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Research 39,
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819-823.
- The Arabidopsis Genome I. 2000. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796-815.
- Wang Z, Gerstein M, Snyder M. 2009. RNA-Seq: A revolutionary tool for transcriptomics. Nature Reviews Genetics 10, 57-63.
______________________________________________
About the Author
Ching Chan is a 2024 Plantae Fellow. Ching’s group is broadly interested in plant-microbe interactions under different environmental conditions and the application of this knowledge to crop improvement. You can find him on X: @ntnuchanlab.

