Single-cell RNA sequencing of developing maize ears facilitates functional analysis and trait candidate gene discovery (Devel. Cell)
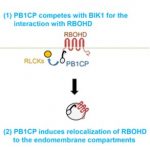
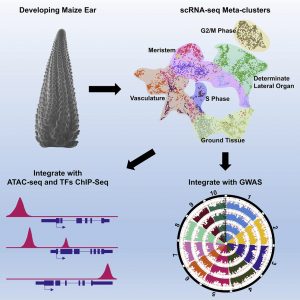 The maize ear has a complex morphology consisting of a series of meristems that are organized into cell types and domains. Xu et al. used single cell RNA sequencing (scRNA-seq) to identify spatial regulators of maize ear development. Over 12,500 individual cells and 28,900 genes were analyzed. 12 meta-clusters were predicted, with inflorescence markers from maize or Arabidopsis used to predict the cell or domain identities of these clusters. The results were validated by in situ hybridization and florescence-activated cell sorting RNA-seq data set. Three applications of the single cell atlas were demonstrated. First, the data predicted redundancy in a maize TPP gene family, which was subsequently validated by multiplex gene-editing using CRISP-Cas9. Second, gene expression is partly controlled by the binding of a transcription factor (TF) to its target genes, therefore targets of a TF should be co-expressed in the same cell types. Based on this underlying assumption, the authors integrated scRNA-seq with ChIP-seq data to build transcriptional regulatory networks. Finally, candidate genes associated with maize ear morphology traits were identified using the association between scRNA-seq marker genes and ear morphology trait SNPs identified in a GWAS panel. The scRNA-seq atlas generated will serve as a valuable resource for maize developmental genetics studies and breeding. (Summary by Toluwase Olukayode @toluxylic) Devel. Cell 10.1016/j.devcel.2020.12.015
The maize ear has a complex morphology consisting of a series of meristems that are organized into cell types and domains. Xu et al. used single cell RNA sequencing (scRNA-seq) to identify spatial regulators of maize ear development. Over 12,500 individual cells and 28,900 genes were analyzed. 12 meta-clusters were predicted, with inflorescence markers from maize or Arabidopsis used to predict the cell or domain identities of these clusters. The results were validated by in situ hybridization and florescence-activated cell sorting RNA-seq data set. Three applications of the single cell atlas were demonstrated. First, the data predicted redundancy in a maize TPP gene family, which was subsequently validated by multiplex gene-editing using CRISP-Cas9. Second, gene expression is partly controlled by the binding of a transcription factor (TF) to its target genes, therefore targets of a TF should be co-expressed in the same cell types. Based on this underlying assumption, the authors integrated scRNA-seq with ChIP-seq data to build transcriptional regulatory networks. Finally, candidate genes associated with maize ear morphology traits were identified using the association between scRNA-seq marker genes and ear morphology trait SNPs identified in a GWAS panel. The scRNA-seq atlas generated will serve as a valuable resource for maize developmental genetics studies and breeding. (Summary by Toluwase Olukayode @toluxylic) Devel. Cell 10.1016/j.devcel.2020.12.015