Know Your Roots: A Transcriptomic Exploration of Key Life History Traits in the Model Lycophyte Selaginella moellendorffii
Land plants (embryophytes) evolved from a freshwater charophycean algal predecessor over 450 million years ago and have since separated into the diversity of plant lineages observed today. A key factor in the expansion of plant life on land was the evolution of rooting structures and a vasculature that enhanced the acquisition of mineral nutrients and provided a critical means for inter-organ communication.
As a sister lineage to spermatophytes (flowering plants and other seed plants), lycophytes are an important system to study the evolution of true roots, vasculature, and the complex biochemistry of natural products related to cell wall biosynthesis. To enable deeper insight into the evolution of plant vasculature, Ferrari et al. (2020) performed extensive RNA-sequencing analysis to resolve the transcriptional landscape of the model lycophyte Selaginella moellendorffii (Selaginella; Figure 1A). The resulting expression atlas captured the transcriptional profiles of all major organs, tissues, and cell types during diurnal (day-night) cycles or upon stress treatments.
 The authors leveraged the “guilt-by-association” principle of co-expression networks to identify and characterize functional gene modules in Selaginella. Importantly, Selaginella expression networks were put into an evolutionary context using in-depth transcriptomic resources available on the CoNekT database (Co-expression Network Toolkit: www.conekt.plant.tools), which includes expression data from a diverse range of extant plant and algal lineages (Proost and Mutwil, 2018; Ferrari et al., 2019). Since lignocellulosic cell walls are a characteristic feature of vascular plants, the authors first investigated the phenylpropanoid/lignin biosynthetic module of Selaginella. An expression network based around the p-coumarate 3-hydroxylase (C3’H) gene involved in the early stages of lignin biosynthesis revealed a Selaginella lignin module that contained enzymes involved in phenylalanine, phenylpropanoid, lignin, and cellulose (cell wall) biosynthesis alongside structural cell wall proteins, polysaccharide-modifying proteins and MYB transcriptional regulator. Similar lignocellulosic modules are present in angiosperms like Arabidopsis thaliana and Brachypodium distachyon (Sibout et al., 2017), suggesting the broad conservation of a vascular plant-specific lignocellulosic module that is at least 400 million years old.
The authors leveraged the “guilt-by-association” principle of co-expression networks to identify and characterize functional gene modules in Selaginella. Importantly, Selaginella expression networks were put into an evolutionary context using in-depth transcriptomic resources available on the CoNekT database (Co-expression Network Toolkit: www.conekt.plant.tools), which includes expression data from a diverse range of extant plant and algal lineages (Proost and Mutwil, 2018; Ferrari et al., 2019). Since lignocellulosic cell walls are a characteristic feature of vascular plants, the authors first investigated the phenylpropanoid/lignin biosynthetic module of Selaginella. An expression network based around the p-coumarate 3-hydroxylase (C3’H) gene involved in the early stages of lignin biosynthesis revealed a Selaginella lignin module that contained enzymes involved in phenylalanine, phenylpropanoid, lignin, and cellulose (cell wall) biosynthesis alongside structural cell wall proteins, polysaccharide-modifying proteins and MYB transcriptional regulator. Similar lignocellulosic modules are present in angiosperms like Arabidopsis thaliana and Brachypodium distachyon (Sibout et al., 2017), suggesting the broad conservation of a vascular plant-specific lignocellulosic module that is at least 400 million years old.
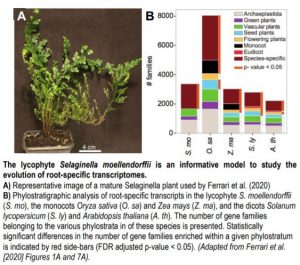
Root transcriptomes are thought to be highly similar despite the pronounced morphological and architectural variation observed across species (Huang and Schiefelbein, 2015). To further examine this phenomenon, Ferrari et al. (2020) identified conserved root-specific transcripts in Selaginella and all available vascular plants in the CoNekT database. Using a phylostratigraphic analysis, which posits that the age of a gene family is based on its earliest appearance in a phylogeny, the authors determined that most root-specific gene families appear first at the species level rather than being conserved across all vascular plants (Figure 1B). This suggests that the evolution of root organs did not coincide with the acquisition and fixation of novel gene families. However, further analysis of the gene families contained within the ‘vascular plant’ phylostratum supports the existence of a core set of root-specific transcripts that are similarly expressed across vascular plants. Expression network analysis of this core set predicted conserved roles in processes ranging from nutrient transport to transcriptional regulation.
Collectively, the Selaginella expression atlas presented by Ferrari and colleagues represents a valuable resource that provides new insight into the evolution of key life history traits in lycophytes. The updated transcriptomes and web-interfaces generated in this study (http://bar.utoronto.ca/efp_selaginella/cgi-bin/efpWeb.cgi) will undoubtedly facilitate future comparative studies aimed at dissecting the core aspects of root biology and secondary metabolism in vascular plants.
Philip Carella
Sainsbury Laboratory
University of Cambridge, Cambridge
ORCID: 0000-0002-5467-7290
REFERENCES
Ferrari C, Proost S, Janowski M, Becker J, Nikoloski Z, Bhattacharya D, Price D, Tohge T, Bar-Even A, Fernie A, Stitt M and Mutwil M (2019). Kingdom-wide comparison reveals the evolution of diurnal gene expression in Archaeplastida. Nat Commun 10: 737.
Ferrari C, Shivhare D, Hansen BO, Pasha A, Esteban E, Provart NJ, Kragler F, Fernie A, Tohge T and Mutwil M (2020) Expression atlas of Selaginella moellendorffii provides insights into the evolution of vasculature, secondary metabolism, and roots. Plant Cell Published January 2020 DOI: https://doi.org/10.1105/tpc.19.00780
Huang L and Schiefelbein J (2015). Conserved Gene Expression Programs in Developing Roots from Diverse Plants. Plant Cell 27: 2119–2132.
Proost S and Mutwil M (2018). CoNekT: an open-source framework for comparative genomic and transcriptomic network analyses. Nucleic Acids Research 46: W133–W140.
Sibout R et al. (2017). Expression atlas and comparative coexpression network analyses reveal important genes involved in the formation of lignified cell wall in Brachypodium distachyon. New Phytol 215: 1009–1025.

