Drawing in the Net: Forty-Five Maize Gene Regulatory Networks from Over 6,000 RNA-Seq Samples
Eukaryotic gene expression is largely governed by transcription factors (TFs), the nuclear proteins that bind to specific DNA sequences and determine when and where genes are turned on. Transcriptional gene regulatory networks (GRNs) that modulate developmental processes and environmental responses consist of TFs and their target genes (nodes), connected by the regulatory interactions among them (edges) (reviewed in Jones and Vandepoele 2020). In recent years, numerous maize (Zea mays) transcriptomic data sets have been generated for various tissues and developmental stages in one or more genotypes, some with stress treatments. These datasets contain a massive amount of information that can be mined for deciphering maize GRNs. However, the plant biology community is currently lacking a unified way to mine this information to understand how plant growth, development, and responses to the environment are regulated.
Zhou et al. (2020) used machine-learning algorithms to infer 45 coexpression-based GRNs from an analysis of 25 maize transcriptome data sets including > 6,000 samples. The authors used three independent approaches to evaluate their validity. First, an analysis of known TF-target interactions based on previously published ChIP-seq data showed that, for 4 of the 6 TFs examined, known targets were significantly enriched in at least one of the putative GRNs. Second, a functional association analysis revealed that all GRNs were significantly enriched for genes that are regulated by the same TF and associated with the same gene ontology term or CornCyc pathway. Finally, an inspection of GRN edges orthologous to known Arabidopsis GRNs showed that 26.5% of those edges are supported by at least one of the 45 maize GRNs. These results suggest that the putative maize GRNs may accurately reflect endogenous gene regulatory processes.
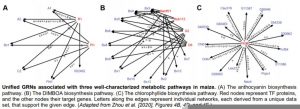 To evaluate consistency amongst the maize GRNs inferred from distinct data sets, the authors examined several metabolic pathways, each with one or more TFs linked to multiple target genes within the pathway. For instance, an analysis of the anthocyanin biosynthesis pathway showed that two of the TFs regulating genes in the pathway were commonly identified in multiple GRNs, but each individual GRN only detected a subset of all the known edges connected to these TFs. Similar phenomena were observed for several other pathways (see figure). This suggests that each GRN inferred from an individual data set uncovers only a subset of the targets of a given TF, and that combining GRNs inferred from multiple data sets may uncover portions of a TF-regulated network in an additive manner.
To evaluate consistency amongst the maize GRNs inferred from distinct data sets, the authors examined several metabolic pathways, each with one or more TFs linked to multiple target genes within the pathway. For instance, an analysis of the anthocyanin biosynthesis pathway showed that two of the TFs regulating genes in the pathway were commonly identified in multiple GRNs, but each individual GRN only detected a subset of all the known edges connected to these TFs. Similar phenomena were observed for several other pathways (see figure). This suggests that each GRN inferred from an individual data set uncovers only a subset of the targets of a given TF, and that combining GRNs inferred from multiple data sets may uncover portions of a TF-regulated network in an additive manner.
Some TFs are known to be differentially expressed in different maize genotypes in certain tissues and/or under certain environmental conditions. An examination of the impact of such TFs on downstream networks showed that, if a TF shows no or minor differential expression in two genotypes (fold change < 2), its putative targets show no or little enrichment of differentially expressed genes. In contrast, for TFs that exhibit higher levels of fold change (> 4), their targets are more likely to be enriched for differentially expressed genes. Therefore, the authors conclude that TFs showing the strongest differential expression between two genotypes might be the most promising candidates for genetic engineering aiming at altering downstream processes.
The authors further explored the association between the maize GRNs and trans-eQTL (expression quantitative trait loci) hotspots, by using published eQTL data sets to investigate whether genes in each hotspot share a common TF regulator. The results indicated that, indeed, in most cases, genes in a given trans-eQTL hotspot tend to likely be regulated by the same TF. By focusing on the statistically best-supported edges in the GRNs that show the strongest enrichment of targets among previously reported trans-eQTL hotspots, the authors identified 68 TFs that co-localize with 74 known trans-eQTL hotspots in the maize genome. Among these 68 TFs, the authors found at least three with well-characterized or putative functions. These results suggest that the GRNs inferred in this study are useful for identifying TFs underlying trans-eQTL hotspots.
The 45 coexpression-based maize GRNs identified, along with the analytical methods developed in this study, represent an outstanding resource for characterizing gene regulatory processes underlying various developmental and stress response processes. Further exploration and utilization of these GRNs show promise to facilitate future breeding and metabolic engineering efforts in maize.
Junpeng Zhan
Donald Danforth Plant Science Center
St. Louis, Missouri, USA
Department of Biology and Institute of Plant and Food Science
Southern University of Science and Technology
Shenzhen, Guangdong, China
[email protected]
ORCID ID: 0000-0001-7353-7608
REFERENCES
Jones, D.M., and Vandepoele, K. (2020). Identification and evolution of gene regulatory networks: insights from comparative studies in plants. Current opinion in plant biology 54: 42-48.
Zhou, P., Li, Z., Magnusson, E., Gomez Cano, F.A., Crisp, P.A., Noshay, J.M., Grotewold, E., Hirsch, C.N., Briggs, S.P., Springer, N.M. (2020). Meta gene regulatory networks in maize highlight functionally relevant regulatory interactions. Plant Cell 32: 10.1105/tpc.20.00080.

