Six Days, Seven Nights: The Transcriptional Speed of Seed Development
The development of the seed is a complex dance of cell division and differentiation, including transcriptional and genomic repatterning. Measuring gene expression by high-throughput RNA-Seq is routine in laboratories, and numerous seed transcriptomes and microarrays have long been published for maize (Grimanelli et al., 2005), but an important caveat is that an RNA-Seq library represents the expressed mRNAs present at a single or few limited points in time. Early seed development occurs particularly fast after pollination, which complicates the identification of genes and gene networks that underlie this crucial developmental stage.
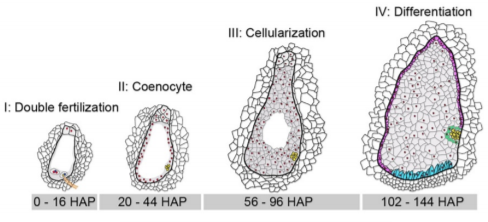
Yi and Gu et al. (2019) elegantly address this question by performing a massive and high-resolution RNA-seq experiment to capture these gene expression changes in the earliest stages of maize B73 seed development after pollination occurs. They painstakingly dissect the small nucellus, a major part of the inner ovule that contains the embryo sac that produces an egg cell for fertilization, at 31 time points from 0 to 144 hours after pollination (HAP). Nearly 3 billion total RNA-seq reads RNA from each of these 31 stages were sequenced and aligned to the maize B73 genome, and then each gene was assigned a normalized expression value at each developmental stage. The authors first took a broad-stroke analysis, performing hierarchical clustering and principal components analysis of gene expression values across the 31 stages. They discovered that the developmental time series clustered into four major stages (I – IV) (see Figure), finely delimiting transitions between double fertilization (Stage I; 0-16 HAP), coenocyte formation (Stage II; 20-44 HAP), cellularization (Stage III; 48-96 HAP), and the initial differentiation of the endosperm (Stage IV; 102-144 HAP). At each of these four major stages, developmental marker genes from a handful of previous studies validated the stage specificity.
By further investigating patterns of gene co-expression within each of these four stages, the authors show that gene networks with distinct functions turn on and off within a matter of hours during seed development, highlighting the power of their high-resolution time-course sampling. For instance, the nearly 4,500 genes and transcription factors expressed during double-fertilization in the 16 hour window of stage I tended to be involved in pollen tube formation and fertilization, involving a suite of MYB transcription factors and known pollen tube genes. Genes expressed in stage II during coenocyte formation tend to be related to DNA replication, transcription, microtubule activity, and nucleosome assembly, plus most of the histone genes, which fits with the active chromatin re-organization and nuclear division typically occurring during this seed stage. Genes expressed in s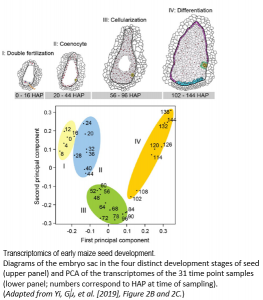 tage III during endosperm cellularization are particularly enriched for functions involving auxin production, response, and transport, in addition to cell membrane formation. Lastly, stage IV genes expressed during endosperm differentiation include a smattering of genes likely involved in cell differentiation and endosperm development.
tage III during endosperm cellularization are particularly enriched for functions involving auxin production, response, and transport, in addition to cell membrane formation. Lastly, stage IV genes expressed during endosperm differentiation include a smattering of genes likely involved in cell differentiation and endosperm development.
Yi and Gu et al. take a powerful next step to identify genes specifically expressed in seed development, integrating publically available data for a smattering of maize reproductive and vegetative tissues. By pairing their high temporal resolution data with several other datasets, they identify nearly 1,000 seed-specific genes, containing nearly 100 transcription factors, which are particularly enriched for expression during stage IV endosperm differentiation and the development of new cell type lineages. This upregulation of seed-specific transcription factors is especially intriguing, and the authors applied gene regulatory network prediction methods to connect transcription factors to their putative target genes.
Perhaps as expected, seed-specific transcription factors tended to regulate seed-specific genes. Moving through each of the 4 major developmental stages identified by the RNA-seq, the authors characterize stage-specific transcription factors that have a high degree of network connectivity, suggesting widespread gene regulation and transcriptional re-patterning that is driven in part by variation in transcription factor networks throughout short spans of developmental time. By focusing on transcription factors that putatively regulate the greatest number of target genes, they find that a handful of MYB, bZIP, and ARF transcription factors may be particularly crucial for modulating gene expression networks during seed development.
Overall, Yi and Gu et al. leverage a staggeringly precise developmental sampling scheme paired with clever informatics to unravel gene regulatory networks, wading through a metaphorical sea of gene candidates to identify genes and transcription factors that regulate seed development. This incredibly robust dataset gives us new insight into the complexities of the maize seed, and will undoubtedly form the foundation for future developmental and functional genomic studies.
REFERENCES
Grimanelli, D., Perotti, E., Ramirez, J., and Leblanc, O. (2005). Timing of the maternal-to-zygotic transition during early seed development in maize. Plant Cell 17: 1061–72.
Yi, F., Gu, W., et al. (2019). High-temporal-resolution transcriptome landscape of early maize seed development.” Plant Cell, March. https://doi.org/10.1105/tpc.18.00961.