Plant Science Research Weekly: June 26th
Review: Plant small heat shock proteins – evolutionary and functional diversity
 Heat shock proteins are rapidly induced by heat treatment and were among the first plant genes and proteins characterized in the early days of molecular biology, nearly 40 years ago. Waters and Vierling review the family of small heat shock proteins (sHSPs), which has been especially amplified in plants and even more so in angiosperms. The authors discuss the domain structure of sHSPS and how they dynamically assemble into multimeric complexes. They also discuss the current model for how sHSPs function, based largely on in vitro studies. Upon heat stress, many proteins unfold, and it is thought that sHSPs interact with these unfolded proteins to promote their interaction with the ATP-dependent chaperones and permit refolding. In vivo studies lag behind in part due to gene redundancy. There is also a role for sHSPs in seeds which may have contribute to desiccation tolerance. (Summary by Mary Williams @PlantTeaching) New Phytol. 10.1111/nph.16536
Heat shock proteins are rapidly induced by heat treatment and were among the first plant genes and proteins characterized in the early days of molecular biology, nearly 40 years ago. Waters and Vierling review the family of small heat shock proteins (sHSPs), which has been especially amplified in plants and even more so in angiosperms. The authors discuss the domain structure of sHSPS and how they dynamically assemble into multimeric complexes. They also discuss the current model for how sHSPs function, based largely on in vitro studies. Upon heat stress, many proteins unfold, and it is thought that sHSPs interact with these unfolded proteins to promote their interaction with the ATP-dependent chaperones and permit refolding. In vivo studies lag behind in part due to gene redundancy. There is also a role for sHSPs in seeds which may have contribute to desiccation tolerance. (Summary by Mary Williams @PlantTeaching) New Phytol. 10.1111/nph.16536
Review. Plant immunity: Danger perception and signaling
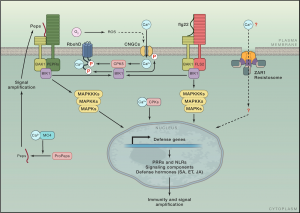 Research from the last three decades has discovered many genes and pathways involved in plant immunity and how they are connected. Here, Zhou and Zhang highlight new research regarding activation and signaling of cell surface pattern recognition receptors (PRRs) and intracellular nucleotide-binding, leucine-rich repeat receptors (NLRs), integration of multiple immunogenic signals to deploy effective defenses, and salicylic acid (SA) biosynthesis and defense gene regulation. Signalling downstream of PRR and NLRs has similarities in their effects, but while PRR responses are transient, they are delayed and prolonged following NLR activation. Transcription of the first enzyme in SA biosynthesis, isochoristmate synthase, is tightly controlled, and SA exerts transcriptional reprogramming by stimulating the transcriptional activation activity of NONEXPRESSOR OF PR GENES 1 (NPR1) while simultaneously inhibiting transcriptional repression by NPR3/NPR4. The interconnectedness of signals from diverse extra- and intra-cellular receptors is truly key to fine tune the defense response against the specific microbe invader. Many questions remain about connections within the plant defense network which will be exciting to see unraveled in the upcoming years. (Summary by Katy Dunning @PlantMomKaty) Cell 10.1016/j.cell.2020.04.028
Research from the last three decades has discovered many genes and pathways involved in plant immunity and how they are connected. Here, Zhou and Zhang highlight new research regarding activation and signaling of cell surface pattern recognition receptors (PRRs) and intracellular nucleotide-binding, leucine-rich repeat receptors (NLRs), integration of multiple immunogenic signals to deploy effective defenses, and salicylic acid (SA) biosynthesis and defense gene regulation. Signalling downstream of PRR and NLRs has similarities in their effects, but while PRR responses are transient, they are delayed and prolonged following NLR activation. Transcription of the first enzyme in SA biosynthesis, isochoristmate synthase, is tightly controlled, and SA exerts transcriptional reprogramming by stimulating the transcriptional activation activity of NONEXPRESSOR OF PR GENES 1 (NPR1) while simultaneously inhibiting transcriptional repression by NPR3/NPR4. The interconnectedness of signals from diverse extra- and intra-cellular receptors is truly key to fine tune the defense response against the specific microbe invader. Many questions remain about connections within the plant defense network which will be exciting to see unraveled in the upcoming years. (Summary by Katy Dunning @PlantMomKaty) Cell 10.1016/j.cell.2020.04.028
Review. The plant microbiome: From ecology to reductionism and beyond
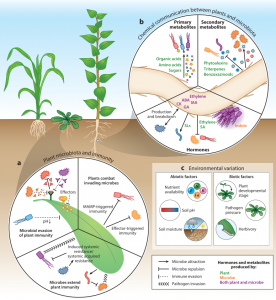 The last two decades have witnessed tremendous progress in our understanding of plant microbiota. Fitzpatrick, Salas-González et al. highlight recent discoveries from culture-dependent and culture-independent approaches and discuss the future path towards integrating these approaches. Culture-independent approaches are census studies of microbial communities in various ecological contexts. Such studies reveal four factors that affect variation in the diversity and composition of plant microbiomes: 1) selection by plants, 2) stochastic variation in growth and death of microbiota members (ecological drift), 3) constant flows of microbes between plants and surrounding environments (dispersal), and 4) evolution within microbial populations. Also, they reveal some bacterial genetic factors important for adaptation to local environments. Culture-dependent approaches include more controlled laboratory experiments that allow a reductionist way of studying mechanisms underlying plant-microbiota interactions. Recent studies have unraveled the roles of plant innate immunity, chemicals produced by plants and microbes, and environmental factors (e.g., biotic and abiotic stresses) in plant microbiome assembly. The authors conclude by providing some examples of how census and reductionist approaches can be integrated towards a unified understanding of the mechanisms and ecological importance of plant-microbiota interactions. (Summary by Tatsuya Nobori @nobolly) Annu. Rev. Microbiol. 10.1146/annurev-micro-022620-014327
The last two decades have witnessed tremendous progress in our understanding of plant microbiota. Fitzpatrick, Salas-González et al. highlight recent discoveries from culture-dependent and culture-independent approaches and discuss the future path towards integrating these approaches. Culture-independent approaches are census studies of microbial communities in various ecological contexts. Such studies reveal four factors that affect variation in the diversity and composition of plant microbiomes: 1) selection by plants, 2) stochastic variation in growth and death of microbiota members (ecological drift), 3) constant flows of microbes between plants and surrounding environments (dispersal), and 4) evolution within microbial populations. Also, they reveal some bacterial genetic factors important for adaptation to local environments. Culture-dependent approaches include more controlled laboratory experiments that allow a reductionist way of studying mechanisms underlying plant-microbiota interactions. Recent studies have unraveled the roles of plant innate immunity, chemicals produced by plants and microbes, and environmental factors (e.g., biotic and abiotic stresses) in plant microbiome assembly. The authors conclude by providing some examples of how census and reductionist approaches can be integrated towards a unified understanding of the mechanisms and ecological importance of plant-microbiota interactions. (Summary by Tatsuya Nobori @nobolly) Annu. Rev. Microbiol. 10.1146/annurev-micro-022620-014327
Convergent loss of plant immune receptors and signaling pathways
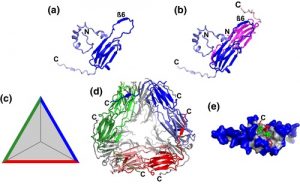
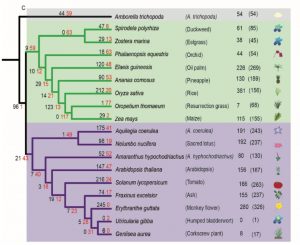 Nucleotide-binding leucine-rich repeat receptors (NLRs) are important components of the plant immune system. They are intracellular receptors that act downstream of the cell-surface receptors, and initiate the so-called effector-triggered immunity (ETI). In most plants, the NLR gene family is large and diverse. In exploring this gene family, Baggs et al. found a small number of angiosperm species that unexpectedly showed independently arising strong contractions of the NLR gene family; these species include duckweed (Spirodela polyrhiza), eelgrass (Zostera marina), corkscrew plant (Genlisea aurea) and humped bladderwort (Utricularia gibba). Intriguingly, these species are adapted to water-saturated marine or bog habitats. Further analysis showed that in these NLR-reduced plants, some of genes that act downstream in the ETI pathway are also reduced or lacking; the authors refer to these genes as ASTREL (AngioSperm Typically-Retained, EDS1-Lost) genes. ASTREL genes include those encoding three proteins that function as a complex, EDS1, PAD4, SAG101. Further study showed that ASTREL genes are normally induced by pathogens or salicylic acid (SA, a hormone that mediates pathogen responses) but suppressed by abscissic acid (ABA, the hormone that mediates drought responses). This study shows that “comparative phylogenomic analysis can fill the gaps in our understanding of complex plant signaling pathways,” and also suggests that the small, fast-growing duckweed S. polyrhiza could be a new model for studies of plant immune signaling. (Summary by Mary Williams @PlantTeaching) Plant Cell 10.1105/tpc.19.00903
Nucleotide-binding leucine-rich repeat receptors (NLRs) are important components of the plant immune system. They are intracellular receptors that act downstream of the cell-surface receptors, and initiate the so-called effector-triggered immunity (ETI). In most plants, the NLR gene family is large and diverse. In exploring this gene family, Baggs et al. found a small number of angiosperm species that unexpectedly showed independently arising strong contractions of the NLR gene family; these species include duckweed (Spirodela polyrhiza), eelgrass (Zostera marina), corkscrew plant (Genlisea aurea) and humped bladderwort (Utricularia gibba). Intriguingly, these species are adapted to water-saturated marine or bog habitats. Further analysis showed that in these NLR-reduced plants, some of genes that act downstream in the ETI pathway are also reduced or lacking; the authors refer to these genes as ASTREL (AngioSperm Typically-Retained, EDS1-Lost) genes. ASTREL genes include those encoding three proteins that function as a complex, EDS1, PAD4, SAG101. Further study showed that ASTREL genes are normally induced by pathogens or salicylic acid (SA, a hormone that mediates pathogen responses) but suppressed by abscissic acid (ABA, the hormone that mediates drought responses). This study shows that “comparative phylogenomic analysis can fill the gaps in our understanding of complex plant signaling pathways,” and also suggests that the small, fast-growing duckweed S. polyrhiza could be a new model for studies of plant immune signaling. (Summary by Mary Williams @PlantTeaching) Plant Cell 10.1105/tpc.19.00903
Multidimensional gene regulatory landscape of a bacterial pathogen in plants
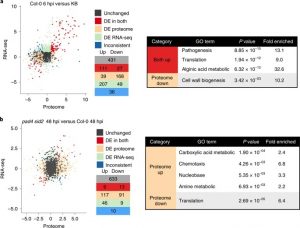 The outcome of a plant-bacteria interaction is determined by the bacterial virulence and plant immune systems. Individually, these systems, and to an extent their interactions, have been well studied. However, certain aspects of their interactions remain elusive, particularly how plant immunity affects bacterial functions. Nobori et al. used an integrated approach of transcriptomics and proteomics analyses to understand how plant immunity modulates bacteria mRNA and protein expression in the Arabidopsis thaliana – Pseudomonas syringae pathosystem. Virulent and an avirulent strains of P. syringae were used. In order to understand the role of the SA pathway and effector-triggered immunity (ETI) on bacterial processes, compromised mutants were included in the analysis. Samples were collected at 6- and 48-hours post infection in order to understand the dynamic changes in mRNA and protein expression at the early and late stage of infection. Comparative analysis showed that mRNA expression moderately correlates with protein expression in all conditions, suggesting that mRNA expression closely reflects protein expression in P. syringae. Further analysis also showed that changes in the bacterial transcriptome are good predictors of changes in the proteome during plant colonization. However, there are a few cases in which mRNA expression changes did not correlate with protein expression; notably, the bacterial cell wall biogenesis-related function was suppressed at the protein but not mRNA level. Finally, previously unknown gene regulatory modules that are important for virulence were also identified. (Summary by Toluwase Olukayode) @toluxylic Nature Plants 10.1038/s41477-020-0690-7
The outcome of a plant-bacteria interaction is determined by the bacterial virulence and plant immune systems. Individually, these systems, and to an extent their interactions, have been well studied. However, certain aspects of their interactions remain elusive, particularly how plant immunity affects bacterial functions. Nobori et al. used an integrated approach of transcriptomics and proteomics analyses to understand how plant immunity modulates bacteria mRNA and protein expression in the Arabidopsis thaliana – Pseudomonas syringae pathosystem. Virulent and an avirulent strains of P. syringae were used. In order to understand the role of the SA pathway and effector-triggered immunity (ETI) on bacterial processes, compromised mutants were included in the analysis. Samples were collected at 6- and 48-hours post infection in order to understand the dynamic changes in mRNA and protein expression at the early and late stage of infection. Comparative analysis showed that mRNA expression moderately correlates with protein expression in all conditions, suggesting that mRNA expression closely reflects protein expression in P. syringae. Further analysis also showed that changes in the bacterial transcriptome are good predictors of changes in the proteome during plant colonization. However, there are a few cases in which mRNA expression changes did not correlate with protein expression; notably, the bacterial cell wall biogenesis-related function was suppressed at the protein but not mRNA level. Finally, previously unknown gene regulatory modules that are important for virulence were also identified. (Summary by Toluwase Olukayode) @toluxylic Nature Plants 10.1038/s41477-020-0690-7
Major impacts of widespread structural variation on gene expression and crop improvement in tomato
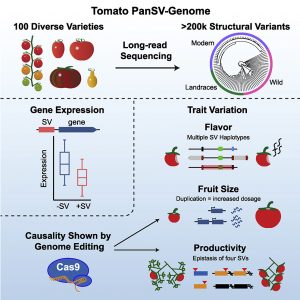 Structural Variants (SVs) are large genomic deletions, insertions, and duplications with underexplored roles in determining plant phenotypes. Recognizing the extent to which SVs define quantitative trait variation was previously constrained by the fact that popular short-read sequencing technologies preferentially characterize smaller variants. However, innovative long-read technologies are facilitating extensive population-level surveys of SVs. Here, Alonge, Wang et al. present panSV, a tomato SV pangenome formed from long-read sequencing of 100 accessions that span phylogenetic diversity. Remarkably, over 238,000 SVs were found, with many shaped by transposons. Across the 100 genomes, 50% of SVs overlap with genes/regulatory sequences, and 95% of genes possess an SV within 5kbp of coding sequences. By employing panSV, 14 new reference genomes, RNA-seq, and CRISPR-Cas9, the authors functionally linked SVs to three major domestication and breeding traits: an unpalatable smoky flavor, a determinant of fruit weight, and jointless fruit pedicels. This study represents the release of the most comprehensive crop SV pangenome, and the abundant resources generated will expedite acknowledgment of the significance of SVs in evolution, domestication, and quantitative trait variation, undoubtedly advancing future crop improvement efforts. (Summary by Caroline Dowling @CarolineD0wling) Cell 10.1016/j.cell.2020.05.021
Structural Variants (SVs) are large genomic deletions, insertions, and duplications with underexplored roles in determining plant phenotypes. Recognizing the extent to which SVs define quantitative trait variation was previously constrained by the fact that popular short-read sequencing technologies preferentially characterize smaller variants. However, innovative long-read technologies are facilitating extensive population-level surveys of SVs. Here, Alonge, Wang et al. present panSV, a tomato SV pangenome formed from long-read sequencing of 100 accessions that span phylogenetic diversity. Remarkably, over 238,000 SVs were found, with many shaped by transposons. Across the 100 genomes, 50% of SVs overlap with genes/regulatory sequences, and 95% of genes possess an SV within 5kbp of coding sequences. By employing panSV, 14 new reference genomes, RNA-seq, and CRISPR-Cas9, the authors functionally linked SVs to three major domestication and breeding traits: an unpalatable smoky flavor, a determinant of fruit weight, and jointless fruit pedicels. This study represents the release of the most comprehensive crop SV pangenome, and the abundant resources generated will expedite acknowledgment of the significance of SVs in evolution, domestication, and quantitative trait variation, undoubtedly advancing future crop improvement efforts. (Summary by Caroline Dowling @CarolineD0wling) Cell 10.1016/j.cell.2020.05.021
The 3′ processing of antisense RNAs physically links to chromatin-based transcriptional control
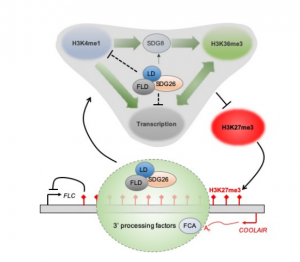 In Arabidopsis, multiple genetic pathways control the floral transition in response to external and internal stimuli. Among these, components of the autonomous pathway promote flowering by negatively regulating the central floral repressor FLOWERING LOCUS C (FLC). Despite the wealth of information on RNA-mediated chromatin silencing of FLC, the molecular link between FLOWERING TIME CONTROL (FCA)- a protein involved in RNA 3′ processing- and FLOWERING LOCUS D (FLD)- a demethylase involved in histone modification- is still missing. In this article, Fang and colleagues undertook a proteomic approach and found physical interaction between the chromatin modifiers FLD, LUMINIDEPENDENS (LD) and SET DOMAIN GROUP 26 (SDG26). Furthermore, Cross-Linked Nuclear Immuno-Precipitation and Mass Spectrometry (CLNIP–MS) analysis showed dynamic interaction of SDG26 not only with FLD-LD but also with different 3′ RNA processing factors including FCA. Additional genetic and molecular studies revealed increased levels of unspliced FLC RNA in higher order mutants, suggesting that the FLD-LD-SDG26 complex is recruited to repress FLC transcription upon RNA binding mediated by FCA. Lastly, quantitative detection of various histone modifications over the FLC gene body in loss of function mutants (fld, ld, sdg26, fca) indicated overaccumulation of histone marks related to transcriptional activation (H3K4me1, H3K36me3) and reduction of histone marks related to gene silencing (H3K27me3), causing late flowering. To conclude, the dynamic interactions between floral activators of the autonomous pathway result in a chromatin state associated with transcriptional inactivation of FLC. (Summary by Michela Osnato @Michela_Osnato) Proc. Natl. Acad. Sci. USA 10.1073/pnas.2007268117
In Arabidopsis, multiple genetic pathways control the floral transition in response to external and internal stimuli. Among these, components of the autonomous pathway promote flowering by negatively regulating the central floral repressor FLOWERING LOCUS C (FLC). Despite the wealth of information on RNA-mediated chromatin silencing of FLC, the molecular link between FLOWERING TIME CONTROL (FCA)- a protein involved in RNA 3′ processing- and FLOWERING LOCUS D (FLD)- a demethylase involved in histone modification- is still missing. In this article, Fang and colleagues undertook a proteomic approach and found physical interaction between the chromatin modifiers FLD, LUMINIDEPENDENS (LD) and SET DOMAIN GROUP 26 (SDG26). Furthermore, Cross-Linked Nuclear Immuno-Precipitation and Mass Spectrometry (CLNIP–MS) analysis showed dynamic interaction of SDG26 not only with FLD-LD but also with different 3′ RNA processing factors including FCA. Additional genetic and molecular studies revealed increased levels of unspliced FLC RNA in higher order mutants, suggesting that the FLD-LD-SDG26 complex is recruited to repress FLC transcription upon RNA binding mediated by FCA. Lastly, quantitative detection of various histone modifications over the FLC gene body in loss of function mutants (fld, ld, sdg26, fca) indicated overaccumulation of histone marks related to transcriptional activation (H3K4me1, H3K36me3) and reduction of histone marks related to gene silencing (H3K27me3), causing late flowering. To conclude, the dynamic interactions between floral activators of the autonomous pathway result in a chromatin state associated with transcriptional inactivation of FLC. (Summary by Michela Osnato @Michela_Osnato) Proc. Natl. Acad. Sci. USA 10.1073/pnas.2007268117
Genome-wide analysis of the U-box E3 ubiquitin ligase enzyme gene family in tomato
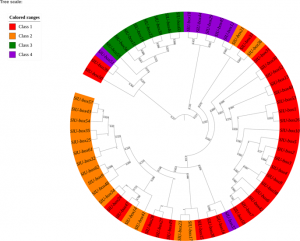 Ubiquitination is a process of adding ubiquitin (a small regulatory protein) to a substrate protein, leading to its degradation. A wide variety of cellular processes necessary for plant growth and development are regulated by ubiquitination. E3 ubiquitin ligases are the largest family of proteins involved in the ubiquitination process and act through transferring target proteins to the degradation pathway. The U-box E3 gene family members are prevalent in plants, however, their evolution and role in promoting development in tomatoes is not well understood. In this study, Sharma and Taganna analyzed the functions of the U-box E3 ubiquitin ligase enzyme gene family in tomato, using a genome-wide study. They identified 62 U-box genes in the tomato genome and reported some of the biochemical and physicochemical properties of the gene family. Through studying gene expression profiles, the authors reported a strong association of the U-box E3 ubiquitin ligases in the development of tomato plant tissues and participation in the stress and hormonal response pathways. This study highlights the role of U-box E3 ligases as genes that can be targeted for the development of crop varieties with increased immunity, yield, and stress tolerance. (Summary by Modesta Abugu @modestannedi). Sci. Reports 10.1038/s41598-020-66553-1
Ubiquitination is a process of adding ubiquitin (a small regulatory protein) to a substrate protein, leading to its degradation. A wide variety of cellular processes necessary for plant growth and development are regulated by ubiquitination. E3 ubiquitin ligases are the largest family of proteins involved in the ubiquitination process and act through transferring target proteins to the degradation pathway. The U-box E3 gene family members are prevalent in plants, however, their evolution and role in promoting development in tomatoes is not well understood. In this study, Sharma and Taganna analyzed the functions of the U-box E3 ubiquitin ligase enzyme gene family in tomato, using a genome-wide study. They identified 62 U-box genes in the tomato genome and reported some of the biochemical and physicochemical properties of the gene family. Through studying gene expression profiles, the authors reported a strong association of the U-box E3 ubiquitin ligases in the development of tomato plant tissues and participation in the stress and hormonal response pathways. This study highlights the role of U-box E3 ligases as genes that can be targeted for the development of crop varieties with increased immunity, yield, and stress tolerance. (Summary by Modesta Abugu @modestannedi). Sci. Reports 10.1038/s41598-020-66553-1
Highly active rubiscos discovered by systematic interrogation of natural sequence diversity
 This is a fascinating and very well-written paper that investigates the diversity of rubisco’s kinetic properties. Rubisco’s relationship with its substrate CO2 is complicated by its relationship with O2, and it has often been suggested that for this reason rubisco is locked into a slow rate of catalysis. The plant enzymes are certainly very slow as compared to other enzymes. Plant enzymes are form I, and assemble as L8S8 complexes, whereas form II and form III enzymes, found in dinoflagellates, bacteria and archea, are complexes of the large subunit only. Here, Davidi et al. queried thousands of genomic sequences to identify about 33,000 diverse rubiscos from across the domains of life and identified more than one hundred that they expressed in E. coli for catalytic assays. They found several that are significantly faster than the plant enzymes. The fastest enzyme they characterized is from a bacterium Gallionella sp. that lives in a very low oxygen environment. (Summary by Mary Williams @PlantTeaching) EMBO J. 10.15252/embj.2019104081
This is a fascinating and very well-written paper that investigates the diversity of rubisco’s kinetic properties. Rubisco’s relationship with its substrate CO2 is complicated by its relationship with O2, and it has often been suggested that for this reason rubisco is locked into a slow rate of catalysis. The plant enzymes are certainly very slow as compared to other enzymes. Plant enzymes are form I, and assemble as L8S8 complexes, whereas form II and form III enzymes, found in dinoflagellates, bacteria and archea, are complexes of the large subunit only. Here, Davidi et al. queried thousands of genomic sequences to identify about 33,000 diverse rubiscos from across the domains of life and identified more than one hundred that they expressed in E. coli for catalytic assays. They found several that are significantly faster than the plant enzymes. The fastest enzyme they characterized is from a bacterium Gallionella sp. that lives in a very low oxygen environment. (Summary by Mary Williams @PlantTeaching) EMBO J. 10.15252/embj.2019104081
Protein complex stoichiometry and expression dynamics of transcription factors modulate stem cell division
 Stem cells are a group of undifferentiated cells that can divide and differentiate to form new organs. In Arabidopsis roots, the quiescent center (QC: the mitotically inactive group of cells) helps regulate the division of surrounding initials and maintain the stem cell fate. What makes the QC different from surrounding stem cells, and how does it regulate the division of its surrounding initials? To address this, Clark et al. studied two transcription factors that form a complex, SCR (SCARECROW) and SHR (SHORTROOT), and determined their roles in the division of QC and Cortex Endodermis Initials (CEI). Using an ordinary differential equation model, the authors were able to predict a higher percentage of the oligomeric state, 1SHR:2SCR and 2SHR:2SCR stoichiometry, in QC compared to CEI. The authors also identified WUSCHEL-RELATED HOMEOBOX5 (WOX5) and SEUSS (SEU) as putative transcriptional regulators of SHR expression. Additionally, the authors also showed how SHR: SCR temporal dynamics can modulate the division of CEI and QC and how SHR homodimer is required for QC division. Overall, the authors in this paper were able to use both mathematical models and experimental data to highlight the role of protein complex stoichiometry in modulating the division of two different cell types in the Arabidopsis root stem cell niche. (Summary by Sunita Pathak @psunita980) Proc. Natl. Acad. Sci. USA 10.1073/pnas.2002166117
Stem cells are a group of undifferentiated cells that can divide and differentiate to form new organs. In Arabidopsis roots, the quiescent center (QC: the mitotically inactive group of cells) helps regulate the division of surrounding initials and maintain the stem cell fate. What makes the QC different from surrounding stem cells, and how does it regulate the division of its surrounding initials? To address this, Clark et al. studied two transcription factors that form a complex, SCR (SCARECROW) and SHR (SHORTROOT), and determined their roles in the division of QC and Cortex Endodermis Initials (CEI). Using an ordinary differential equation model, the authors were able to predict a higher percentage of the oligomeric state, 1SHR:2SCR and 2SHR:2SCR stoichiometry, in QC compared to CEI. The authors also identified WUSCHEL-RELATED HOMEOBOX5 (WOX5) and SEUSS (SEU) as putative transcriptional regulators of SHR expression. Additionally, the authors also showed how SHR: SCR temporal dynamics can modulate the division of CEI and QC and how SHR homodimer is required for QC division. Overall, the authors in this paper were able to use both mathematical models and experimental data to highlight the role of protein complex stoichiometry in modulating the division of two different cell types in the Arabidopsis root stem cell niche. (Summary by Sunita Pathak @psunita980) Proc. Natl. Acad. Sci. USA 10.1073/pnas.2002166117
Macroevolutionary patterns in seed component mass and different evolutionary trajectories across seed desiccation responses ($)
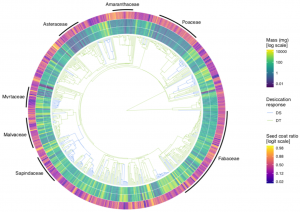 Seeds are made out of two functionally distinct components: the seed coat and the seed reserves (which include endosperm and embryo). It is known that the relative contribution of these parts to the total seed mass varies greatly among species. Still, little is known about the evolution of each component’s mass and proportion. In this fascinating paper, Chen et al. provide the first assessment of these traits’ evolutionary trajectories, with a particular interest in testing whether these trajectories change between desiccation-tolerant and desiccation-sensitive seeds. Using comparative phylogenetic analysis, the authors found that seed reserve mass had higher evolutionary rates than seed coat mass. Therefore, they suggest that the allocation of resources between the two seed components is mainly driven by changes in seed reserves. Overall, this behavior is independent of seed desiccation responses. However, desiccation-sensitive species had relatively more seed reserve mass. Given this, the authors discuss that the evolution of seed reserve mass is key to understanding the appearance of desiccation sensitivity in seeds. (Summary by Carlos A. Ordóñez-Parra @caordonezparra) New Phytol. 10.1111/nph.16706
Seeds are made out of two functionally distinct components: the seed coat and the seed reserves (which include endosperm and embryo). It is known that the relative contribution of these parts to the total seed mass varies greatly among species. Still, little is known about the evolution of each component’s mass and proportion. In this fascinating paper, Chen et al. provide the first assessment of these traits’ evolutionary trajectories, with a particular interest in testing whether these trajectories change between desiccation-tolerant and desiccation-sensitive seeds. Using comparative phylogenetic analysis, the authors found that seed reserve mass had higher evolutionary rates than seed coat mass. Therefore, they suggest that the allocation of resources between the two seed components is mainly driven by changes in seed reserves. Overall, this behavior is independent of seed desiccation responses. However, desiccation-sensitive species had relatively more seed reserve mass. Given this, the authors discuss that the evolution of seed reserve mass is key to understanding the appearance of desiccation sensitivity in seeds. (Summary by Carlos A. Ordóñez-Parra @caordonezparra) New Phytol. 10.1111/nph.16706