Plant Science Research Weekly: February 15th
A comparison of the EU regulatory approach to directed mutagenesis with that of other jurisdictions, consequences for international trade and potential steps forward
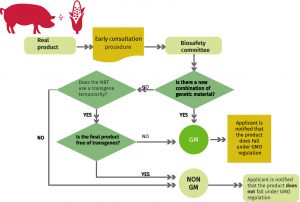 The seed sector, and particularly plant breeders, are responsible for providing farmers with new plant varieties able to overcome challenges such as climate change, water restrictions or plant pests. In order to do their job, breeders have a collection of tools at their disposal. Among these are traditional randomly induced mutagenesis and, since the 1970s, recombinant nucleic acid technology that produces the so-called genetically modified organisms (GMOs) also commonly known as transgenic organisms. In recent years they also have genome editing techniques such as CRISPR/CAS9 that enable precise and beneficial mutations without the costs and extra workload that come with arbitrarily induced mutagenesis. Each tool has its pros and cons. A recent ruling from the European Court of Justice might hinder the global development and trade of plant varieties using newly established CRISPR/CAS9-like technologies. In order to shed light on the current regulatory frameworks on directed mutagenesis, an international team of experts led by Dennis Erikson has produced an in-depth comparison between the EU situation and the existing legislation at seven other countries (Argentina, Australia, Brazil, Canada, Chile, Colombia and the United States in North America) (Summary by Isabel Mendoza) New Phytol. 10.1111/nph.15627
The seed sector, and particularly plant breeders, are responsible for providing farmers with new plant varieties able to overcome challenges such as climate change, water restrictions or plant pests. In order to do their job, breeders have a collection of tools at their disposal. Among these are traditional randomly induced mutagenesis and, since the 1970s, recombinant nucleic acid technology that produces the so-called genetically modified organisms (GMOs) also commonly known as transgenic organisms. In recent years they also have genome editing techniques such as CRISPR/CAS9 that enable precise and beneficial mutations without the costs and extra workload that come with arbitrarily induced mutagenesis. Each tool has its pros and cons. A recent ruling from the European Court of Justice might hinder the global development and trade of plant varieties using newly established CRISPR/CAS9-like technologies. In order to shed light on the current regulatory frameworks on directed mutagenesis, an international team of experts led by Dennis Erikson has produced an in-depth comparison between the EU situation and the existing legislation at seven other countries (Argentina, Australia, Brazil, Canada, Chile, Colombia and the United States in North America) (Summary by Isabel Mendoza) New Phytol. 10.1111/nph.15627
Single-cell RNA sequencing resolves molecular relationships among individual plant cells
![]() Single-cell RNA sequencing (scRNA-seq) has transformed our understanding of gene expression within and amongst individual cells. These techniques have been applied extensively to animal cell populations to discover the development of specific cell lines and identify rare cell types but are not commonly used in plant biology. The study here presents high-throughput scRNA-seq data for Arabidopsis thaliana seedling roots. Analysis of single cell transcriptomic profiles reveals that gene expression from all root tissues are identified improving insights into cell-type differentiation. Further work demonstrates that the approach has the power to recognise rare cell types and differentiate developmental signatures between wild and mutant plants. The paper highlights the untapped potential of scRNA-seq to provide gene expression at single cell resolution which could be applied to other species and plant organs (Summary by Alex Bowles) Plant Physiology 10.1104/pp.18.01482.
Single-cell RNA sequencing (scRNA-seq) has transformed our understanding of gene expression within and amongst individual cells. These techniques have been applied extensively to animal cell populations to discover the development of specific cell lines and identify rare cell types but are not commonly used in plant biology. The study here presents high-throughput scRNA-seq data for Arabidopsis thaliana seedling roots. Analysis of single cell transcriptomic profiles reveals that gene expression from all root tissues are identified improving insights into cell-type differentiation. Further work demonstrates that the approach has the power to recognise rare cell types and differentiate developmental signatures between wild and mutant plants. The paper highlights the untapped potential of scRNA-seq to provide gene expression at single cell resolution which could be applied to other species and plant organs (Summary by Alex Bowles) Plant Physiology 10.1104/pp.18.01482.
The genome of broomcorn millet: The fourth millet to be sequenced
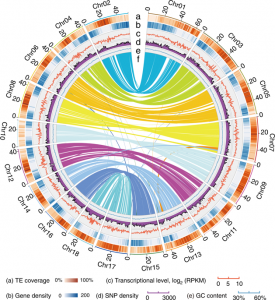 Whole genome sequencing (WGS) unlocks the repertoire of genes encoded within the genome. From a structural genomics aspect, the sequence data enables the development of molecular markers and maps, and identification of novel QTLs/genes/alleles regulating the trait-of-interest. From a functional genomics perspective, the sequence information provides direct access to the genes responsible for the trait-of-interest, which could be used in developing transgenics. WGS has been popular among the ‘known’ crops including rice, wheat, barley, etc. However, a predominant category of plants remains ‘understudied’: these include millets, tubers, legumes and leafy vegetables. Among millets, the genomes of three species have been sequenced, namely, foxtail, pearl and finger millet. Now, broomcorn millet enters the genome club, and of note, broomcorn is the first minor millet to get its genome sequenced. Illumina short-read coupled with Pac-Bio long-read sequencing of this allotetraploid crop (2n = 4X= 36; ~923 Mb) provided the information about 55930 protein-coding genes and 339 microRNA genes present in the genome. In addition, 132 individuals from an F6 population of recombinant inbred lines (RIL) were also sequenced, and a high-density genetic map was constructed using 221787 SNP markers. This information will expedite structural and functional genomics studies in broomcorn millet, and also assist crop improvement programs. (Summary by Muthamilarasan Mehanathan). Nature Comms. 10.1038/s41467-019-08409-5
Whole genome sequencing (WGS) unlocks the repertoire of genes encoded within the genome. From a structural genomics aspect, the sequence data enables the development of molecular markers and maps, and identification of novel QTLs/genes/alleles regulating the trait-of-interest. From a functional genomics perspective, the sequence information provides direct access to the genes responsible for the trait-of-interest, which could be used in developing transgenics. WGS has been popular among the ‘known’ crops including rice, wheat, barley, etc. However, a predominant category of plants remains ‘understudied’: these include millets, tubers, legumes and leafy vegetables. Among millets, the genomes of three species have been sequenced, namely, foxtail, pearl and finger millet. Now, broomcorn millet enters the genome club, and of note, broomcorn is the first minor millet to get its genome sequenced. Illumina short-read coupled with Pac-Bio long-read sequencing of this allotetraploid crop (2n = 4X= 36; ~923 Mb) provided the information about 55930 protein-coding genes and 339 microRNA genes present in the genome. In addition, 132 individuals from an F6 population of recombinant inbred lines (RIL) were also sequenced, and a high-density genetic map was constructed using 221787 SNP markers. This information will expedite structural and functional genomics studies in broomcorn millet, and also assist crop improvement programs. (Summary by Muthamilarasan Mehanathan). Nature Comms. 10.1038/s41467-019-08409-5
Tapping into the genetic diversity of wild crops for engineering disease resistance
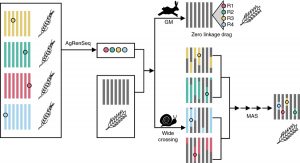 Lack of sequence information makes harnessing the diversity of disease resistance (R) genes in wild crops highly challenging. Arora and Steuernagel et al. report AgRenSeq, a reference genome-free technique, for rapid cloning of nucleotide binding/Leucine-rich repeat (NLR) resistance genes from wild crops. Using R gene enrichment sequencing, the authors captured thousands of NLR-specific contigs from 151 distinct accessions of Aegilops taushii, a wild relative of bread wheat. Based on k-mer based association mapping, they identified four wheat stem rust resistance genes from the captured contigs. AgRenSeq thus provides a powerful technique for discovering rare, uncharacterized R genes from genetically diverse accessions of any crop and improving disease resistance in its domesticated counterparts. (Summary by Saima Shahid) Nature Biotech. 10.1038/s41587-018-0007-9.
Lack of sequence information makes harnessing the diversity of disease resistance (R) genes in wild crops highly challenging. Arora and Steuernagel et al. report AgRenSeq, a reference genome-free technique, for rapid cloning of nucleotide binding/Leucine-rich repeat (NLR) resistance genes from wild crops. Using R gene enrichment sequencing, the authors captured thousands of NLR-specific contigs from 151 distinct accessions of Aegilops taushii, a wild relative of bread wheat. Based on k-mer based association mapping, they identified four wheat stem rust resistance genes from the captured contigs. AgRenSeq thus provides a powerful technique for discovering rare, uncharacterized R genes from genetically diverse accessions of any crop and improving disease resistance in its domesticated counterparts. (Summary by Saima Shahid) Nature Biotech. 10.1038/s41587-018-0007-9.
Distinct characteristics of genes associated with phenome-wide variation in maize (Zea mays)
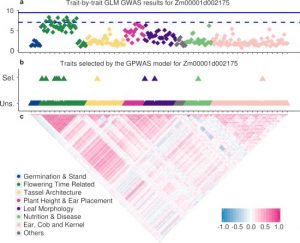 High-throughput plant phenotyping is growing rapidly and enables the collection of dozens or hundreds of traits of the same plant genotype efficiently. The development of this technology expands the diversity of plant phenotypes and brings an opportunity for reexamining the connections between genotype and phenotype from a novel perspective. The widely adopted genome-wide association study (GWAS) provides meaningful evidence for gene function exploration. In this study, the authors proposed a Genome-Phenome Wide Association Study (GPWAS) model that couples hundreds of commonly accepted agronomic traits to explain variants in the gene level of maize. Based on gene features from evolution, expression, structure variation and gene annotation, genes identified by GPWAS tends to have closer distance with classical maize mutants than genes in background or identified by conventional GWAS models. This study shows the potential for utilizing high-dimensional phenotypes in identifying desirably functional candidates from a large number of annotated genes. (Summary by Zhikai Liang) bioRxiv: 10.1101/534503v2
High-throughput plant phenotyping is growing rapidly and enables the collection of dozens or hundreds of traits of the same plant genotype efficiently. The development of this technology expands the diversity of plant phenotypes and brings an opportunity for reexamining the connections between genotype and phenotype from a novel perspective. The widely adopted genome-wide association study (GWAS) provides meaningful evidence for gene function exploration. In this study, the authors proposed a Genome-Phenome Wide Association Study (GPWAS) model that couples hundreds of commonly accepted agronomic traits to explain variants in the gene level of maize. Based on gene features from evolution, expression, structure variation and gene annotation, genes identified by GPWAS tends to have closer distance with classical maize mutants than genes in background or identified by conventional GWAS models. This study shows the potential for utilizing high-dimensional phenotypes in identifying desirably functional candidates from a large number of annotated genes. (Summary by Zhikai Liang) bioRxiv: 10.1101/534503v2
SOL1 and SOL2 regulate fate transition and cell division in the Arabidopsis stomatal lineage ($)
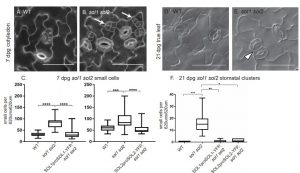 Stomata and pavement cell development in the leaves occur through programmed asymmetric cell division. In this paper the authors demonstrated the role of two CHC domain proteins, SOL1 and SOL2 that were identified previously by the same group to be downstream targets of a bHLH transcriptional factor SPEECHLESS. The two genes are close homologs of TSO1 which is known to play crucial role in floral meristem maintenance. SOL1 is expressed in the early stages of stomatal lineage cells ahead of cell division and SOL2 in the meristemoid and GMC as illustrated by co-expression with lineage specific markers. The double loss of function mutant sol1sol2 causes development of stomatal pairs and increased small cell phenotype which is rescued by gene complementation. Together these results showed the function of two important components in stomatal development. (Summary by Suresh Damodaran) Development
Stomata and pavement cell development in the leaves occur through programmed asymmetric cell division. In this paper the authors demonstrated the role of two CHC domain proteins, SOL1 and SOL2 that were identified previously by the same group to be downstream targets of a bHLH transcriptional factor SPEECHLESS. The two genes are close homologs of TSO1 which is known to play crucial role in floral meristem maintenance. SOL1 is expressed in the early stages of stomatal lineage cells ahead of cell division and SOL2 in the meristemoid and GMC as illustrated by co-expression with lineage specific markers. The double loss of function mutant sol1sol2 causes development of stomatal pairs and increased small cell phenotype which is rescued by gene complementation. Together these results showed the function of two important components in stomatal development. (Summary by Suresh Damodaran) Development
KAI2 regulates root and root hair development by modulating auxin distribution
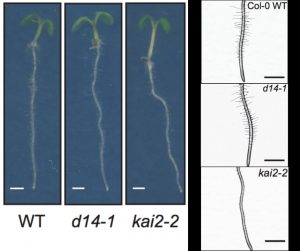 To optimize growth, plant development is regulated by environmental information. Root morphology is adaptable to stimuli due to a network of phytohormone signalling pathways. Here Villaecija et al. studied the roles of strigolactones (SL) and karrikins (KL) signalling in controlling root and root hair development in Arabidopsis. SLs and KLs are similar in size and structure, and both act through the same Fbox protein MAX2. By using mutants affected in SL biosynthesis and SL and KL receptors (D14 and KAI2) this study shows, conversely to earlier studies, that SL signalling has minor effects while KL signalling is a key regulator of root hair development. KL regulates auxin distribution through the concentration of the protein PIN7 in the root tip. Furthermore, this study suggest KL signalling alone suppresses root skewing and modulates root straightness, growth direction and diameter. This work helps to understand important signalling pathways which may lead to plant development and tune growth for agriculture improvement. (Summary by Ana Valladares) bioRxiv https://doi.org/10.1101/539734
To optimize growth, plant development is regulated by environmental information. Root morphology is adaptable to stimuli due to a network of phytohormone signalling pathways. Here Villaecija et al. studied the roles of strigolactones (SL) and karrikins (KL) signalling in controlling root and root hair development in Arabidopsis. SLs and KLs are similar in size and structure, and both act through the same Fbox protein MAX2. By using mutants affected in SL biosynthesis and SL and KL receptors (D14 and KAI2) this study shows, conversely to earlier studies, that SL signalling has minor effects while KL signalling is a key regulator of root hair development. KL regulates auxin distribution through the concentration of the protein PIN7 in the root tip. Furthermore, this study suggest KL signalling alone suppresses root skewing and modulates root straightness, growth direction and diameter. This work helps to understand important signalling pathways which may lead to plant development and tune growth for agriculture improvement. (Summary by Ana Valladares) bioRxiv https://doi.org/10.1101/539734
Light regulates plant alternative splicing through the control of transcriptional elongation
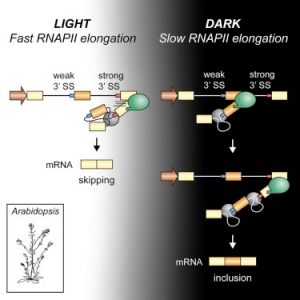 Light perception in plants generates a plethora of gene expression changes. Among them, it has been shown that light affects alternative splicing mediated by a chloroplast retrograde signal. Here, Godoy Herz et al. explore the different possible mechanisms possible involved in this regulation. Using either pharmacological inhibitors or an Arabidopsis mutant in histone deacetylation, they found similar splicing patterns to those observed in light, but this epigenetic mark does not explain the mechanism involved. Next, mutations or drugs affecting RNA pol II elongation eliminate or reduce the effect of light on splicing, respectively. Using different techniques, the authors also show that RNA pol II elongation is affected in dark conditions. These findings are in accordance with the Kinetic Model of coupling between transcription and alternative splicing first described in mammalian cells. This work shows how this mechanism is important for environmental responses in biologically relevant conditions. However, it is still necessary to know the basis for the connection between the chloroplast retrograde signal and transcript elongation. (Summary by Facundo Romani) Mol. Plant 10.1016/j.molcel.2018.12.005
Light perception in plants generates a plethora of gene expression changes. Among them, it has been shown that light affects alternative splicing mediated by a chloroplast retrograde signal. Here, Godoy Herz et al. explore the different possible mechanisms possible involved in this regulation. Using either pharmacological inhibitors or an Arabidopsis mutant in histone deacetylation, they found similar splicing patterns to those observed in light, but this epigenetic mark does not explain the mechanism involved. Next, mutations or drugs affecting RNA pol II elongation eliminate or reduce the effect of light on splicing, respectively. Using different techniques, the authors also show that RNA pol II elongation is affected in dark conditions. These findings are in accordance with the Kinetic Model of coupling between transcription and alternative splicing first described in mammalian cells. This work shows how this mechanism is important for environmental responses in biologically relevant conditions. However, it is still necessary to know the basis for the connection between the chloroplast retrograde signal and transcript elongation. (Summary by Facundo Romani) Mol. Plant 10.1016/j.molcel.2018.12.005
Chloroplast status can affect shade avoidance in plants ($)
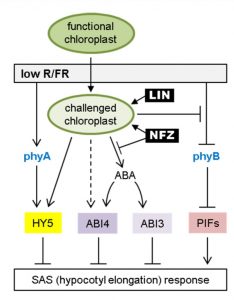 For optimal photosynthetic performance, plants utilize multiple strategies to compete with their neighbors. Especially when light supply is limited in the canopy, plants promote their own elongation to overgrow neighboring plants. This occurs as a response to a low ratio of Red to Far-Red light (R/FR), in a process called the Shade Avoidance Syndrome (SAS). Recently Ortiz-Alcaide et al. revealed the essential role of chloroplast function in SAS, without which the stem of Arabidopsis cannot elongate in canopy shade. More importantly, they found that low R/FR may trigger a feedback mechanism in SAS via inhibiting phyB deactivation, increasing HY5 accumulation and promoting ABA signaling to prevent excessive elongation. This is an elegant study of the interaction between light and retrograde pathway that shows that chloroplasts are far more than a photosynthesis platform, but are also important for light sensing and signaling. (Summary by Nanxunq Qin). Plant Cell 10.1105/tpc.18.00617
For optimal photosynthetic performance, plants utilize multiple strategies to compete with their neighbors. Especially when light supply is limited in the canopy, plants promote their own elongation to overgrow neighboring plants. This occurs as a response to a low ratio of Red to Far-Red light (R/FR), in a process called the Shade Avoidance Syndrome (SAS). Recently Ortiz-Alcaide et al. revealed the essential role of chloroplast function in SAS, without which the stem of Arabidopsis cannot elongate in canopy shade. More importantly, they found that low R/FR may trigger a feedback mechanism in SAS via inhibiting phyB deactivation, increasing HY5 accumulation and promoting ABA signaling to prevent excessive elongation. This is an elegant study of the interaction between light and retrograde pathway that shows that chloroplasts are far more than a photosynthesis platform, but are also important for light sensing and signaling. (Summary by Nanxunq Qin). Plant Cell 10.1105/tpc.18.00617
A regulatory circuit conferring varied flowering response to cold in annual and perennial plants ($)
 Regulation of flowering in time and space in perennials like Arabis alpina involves two systems. One uses orthologs to FLOWERING LOCUS C (FLC) from A. thaliana; in A. alpine is called PERPETUAL FLOWERING 1 (PEP1) and its repression enables flowering after vernalization. The other system uses microRNA156 (miR156), which allows the flowering response after vernalization only in older meristems. It has been shown that miR156 targets SQUAMOSA PROMOTER BINDING PROTEIN LIKE -15 (SPL-15). Hyun and collaborators, using CRISPR/Cas9 to inactivate SPL-15, show how the two systems are linked in perennials through SPL-15, acting only in older shoots and branches after vernalization. To test SPL-15’s role in annuals, lines from an interspecific cross between A. alpina and annual Arabis montbretiana showed that the flowering response occurred in young shoots after vernalization through photoperiod and without SPL-15 mediation. (Summary by Cecilia Vasquez-Robinet) Science 10.1126/science.aau8197
Regulation of flowering in time and space in perennials like Arabis alpina involves two systems. One uses orthologs to FLOWERING LOCUS C (FLC) from A. thaliana; in A. alpine is called PERPETUAL FLOWERING 1 (PEP1) and its repression enables flowering after vernalization. The other system uses microRNA156 (miR156), which allows the flowering response after vernalization only in older meristems. It has been shown that miR156 targets SQUAMOSA PROMOTER BINDING PROTEIN LIKE -15 (SPL-15). Hyun and collaborators, using CRISPR/Cas9 to inactivate SPL-15, show how the two systems are linked in perennials through SPL-15, acting only in older shoots and branches after vernalization. To test SPL-15’s role in annuals, lines from an interspecific cross between A. alpina and annual Arabis montbretiana showed that the flowering response occurred in young shoots after vernalization through photoperiod and without SPL-15 mediation. (Summary by Cecilia Vasquez-Robinet) Science 10.1126/science.aau8197
Seed size is regulated by siRNAs from Arabidopsis maternal tissue in a spatial-temporal manner ($)
 Studying seed development is important for understanding plant evolution and engineering food production. Previous discoveries have shown that maternal small interfering RNAs (siRNAs), which induce RNA-directed DNA methylation (RdDM) through NRPD1-mediated pathway, regulate seed development in Arabidopsis. However, it was ambiguous whether the maternal expression of these NRPD1-siRNAs is from the seed coat (maternal) or from the endosperm (uniparentally expressed alleles). Here, Kirkbride et al. used laser-capture microdissection (LCM) to separate small RNA in the endosperm and seed coat and investigate their biological effects. This study shows that the spatial-temporal expression of specific AGAMOUS-LIKE (AGL) transcription factors is regulated by maternal siRNAs, and the ON/OFF of these imprinted genes in the endosperm changes seed size. These discoveries provide a potential strategy to optimize seed production and crop yields. (Summary by Jin Liao) Proc. Natl. Acad. Sci. USA 10.1073/pnas.1807621116
Studying seed development is important for understanding plant evolution and engineering food production. Previous discoveries have shown that maternal small interfering RNAs (siRNAs), which induce RNA-directed DNA methylation (RdDM) through NRPD1-mediated pathway, regulate seed development in Arabidopsis. However, it was ambiguous whether the maternal expression of these NRPD1-siRNAs is from the seed coat (maternal) or from the endosperm (uniparentally expressed alleles). Here, Kirkbride et al. used laser-capture microdissection (LCM) to separate small RNA in the endosperm and seed coat and investigate their biological effects. This study shows that the spatial-temporal expression of specific AGAMOUS-LIKE (AGL) transcription factors is regulated by maternal siRNAs, and the ON/OFF of these imprinted genes in the endosperm changes seed size. These discoveries provide a potential strategy to optimize seed production and crop yields. (Summary by Jin Liao) Proc. Natl. Acad. Sci. USA 10.1073/pnas.1807621116
Lateral inhibition by a peptide hormone-receptor cascade during Arabidopsis lateral root founder cell formation
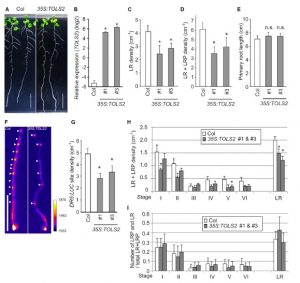
The plant hormone auxin acts as a general coordinator of growth and development, transferring information over both long and short ranges. Among other roles, it promotes the formation of the founder cells in roots and it triggers the position of the lateral roots (LR). LR founder cells are determined in the oscillation zone at the root apex for the LR formation, but sometimes two pairs are formed in close proximaty yet only one persists. Toyokura et al. describe here how a peptide hormone-receptor cascade plays an essential role in the proper LR spacing. In fact, auxin affects LR spacing by inducing expression of the secreted peptide TOLS2/PIPL3 in the LR founder cells, which negatively regulates LR initiation through a lateral inhibition mechanism controlled by the signalling cascade TOLS2-RLK-PUCHI in a non-cell autonomous manner. (Summary by Francesca Resentini) Devel. Cell 10.1016/j.devcel.2018.11.031
The Arabidopsis thaliana pan-NLRome; towards exploring NLR diversity in plants
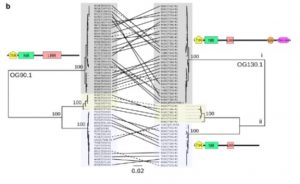 Nucleotide-binding leucine-rich repeat (NLRs) receptors are involved in the intracellular recognition of pathogen effectors. Hundreds of genes encode these NLRs, and the genes are highly polymorphic. So far, a limited number of NLRs were characterized, and three mechanisms of effector recognition have been reported. However, there could be additional mechanisms that operate in the plants which can be studied only if the complete NLRome is decoded. NLR sequence enrichment and long-read sequencing (SMRT RenSeq) of 65 diverse Arabidopsis thaliana accessions identified 13167 NLR genes. Approximately 5% of the genes had at least one additional integrated protein domain (ID). Several IDs were found to be novel as compared to Col-0 reference, and this exposes the largely unexplored repertoires of NLR-IDs in Arabidopsis. Functional characterization of these IDs would provide insights into the different mechanisms underlying the recognition of effectors. The work thus provides a blueprint for investigating the NLRome in other plant species. (Summary by Muthamilarasan Mehanathan). bioRxiv 10.1101/537001v1
Nucleotide-binding leucine-rich repeat (NLRs) receptors are involved in the intracellular recognition of pathogen effectors. Hundreds of genes encode these NLRs, and the genes are highly polymorphic. So far, a limited number of NLRs were characterized, and three mechanisms of effector recognition have been reported. However, there could be additional mechanisms that operate in the plants which can be studied only if the complete NLRome is decoded. NLR sequence enrichment and long-read sequencing (SMRT RenSeq) of 65 diverse Arabidopsis thaliana accessions identified 13167 NLR genes. Approximately 5% of the genes had at least one additional integrated protein domain (ID). Several IDs were found to be novel as compared to Col-0 reference, and this exposes the largely unexplored repertoires of NLR-IDs in Arabidopsis. Functional characterization of these IDs would provide insights into the different mechanisms underlying the recognition of effectors. The work thus provides a blueprint for investigating the NLRome in other plant species. (Summary by Muthamilarasan Mehanathan). bioRxiv 10.1101/537001v1
Cyanobacterial antimetabolite 7-deoxy-sedoheptulose blocks the shikimate pathway to inhibit the growth of prototrophic organisms
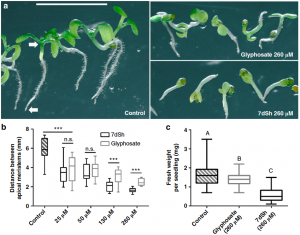 The shikimate pathway synthesizes aromatic amino acids in microorganisms and plants, and its absence in animals makes the shikimate pathway a common target for the development of herbicides. Brilisauer et al. 2019 isolated a novel compound from the cyanobacterium Synechococcus elongatus—7-deoxy-sedoheptulose (7dSH)—that blocks 3-deoxy-D-arabino-heptulosonate 7-phospate (DAHP) from binding to 3-dehydroquinate (DHQ) synthase, a key step in the shikimate pathway. Treatment of Anabaena variabilis (a cyanobacterium), Saccharomyces cerevisiae, and Arabidopsis thaliana with 7dSH inhibited their growth, and in the case of Arabidopsis was more potent than equivalent treatment with glyphosate. This study broadens our understanding of cyanobacterial defense and identifies a compound with the potential for future agricultural application. (Summary by Nathan Scinto-Madonich) Nature Comms 10.1038/s41467-019-08476-8
The shikimate pathway synthesizes aromatic amino acids in microorganisms and plants, and its absence in animals makes the shikimate pathway a common target for the development of herbicides. Brilisauer et al. 2019 isolated a novel compound from the cyanobacterium Synechococcus elongatus—7-deoxy-sedoheptulose (7dSH)—that blocks 3-deoxy-D-arabino-heptulosonate 7-phospate (DAHP) from binding to 3-dehydroquinate (DHQ) synthase, a key step in the shikimate pathway. Treatment of Anabaena variabilis (a cyanobacterium), Saccharomyces cerevisiae, and Arabidopsis thaliana with 7dSH inhibited their growth, and in the case of Arabidopsis was more potent than equivalent treatment with glyphosate. This study broadens our understanding of cyanobacterial defense and identifies a compound with the potential for future agricultural application. (Summary by Nathan Scinto-Madonich) Nature Comms 10.1038/s41467-019-08476-8

