A Roadmap Toward Large-Scale Genome Editing in Crops
If the planet does not turn to ash before 2050, farmers will need to produce twice as much on roughly the same available arable land to feed the ever-growing world population. But since the Green Revolution of the 1950s and 1960s, progress in crop yield has been incremental. And for good reason: most agronomically-relevant traits in crops are polygenic in nature. And although candidate genes for higher yield and productivity abound from the concerted efforts of genetic screens and the mining of natural variation resources by genome-wide association studies (GWAS) and quantitative trait loci (QTLs), the confirmation of a specific gene as the cause of an underlying QTL (or any variant, natural or induced), can be a real beast. Here, Liu et al (2020) illustrate beautifully how new tools can be harnessed to make brute-force approaches more manageable for gene discovery and validation.
A major bottlenecks at the end of a mapping project to a mutant or QTL of interest has typically been 1) picking the gene to focus your efforts on within the mapping interval, and 2) demonstrating causality for the phenotype under consideration. Genome sequence gazing, in silico expression profiling and phylogenetics can help you choose a candidate gene, but what if you pick wrong? What if there is no phenotype, or, perhaps worse, there is a phenotype but it has nothing to do with what you were looking for?
 Well, Liu et al. (2020) propose something new here: why choose? So you have ten genes in your interval; target them all, thanks to genome editing via the CRISPR-Cas9. Since its introduction in 2012, this technique (which stands for Clustered Regularly Interspaced Short Palindromic Repeats and CRISPR-Associated nuclease), has truly revolutionized genetics. Animals studies were very quick to develop libraries of guide RNAs (sgRNAs) that Cas9 needs to target a gene of interest. Cas9-assisted genome editing works in crops too, including maize, but few studies have been brave enough to scale up the analysis by generating and analyzing thousands of transformants. Liu et al. found the courage to step up to the challenge.
Well, Liu et al. (2020) propose something new here: why choose? So you have ten genes in your interval; target them all, thanks to genome editing via the CRISPR-Cas9. Since its introduction in 2012, this technique (which stands for Clustered Regularly Interspaced Short Palindromic Repeats and CRISPR-Associated nuclease), has truly revolutionized genetics. Animals studies were very quick to develop libraries of guide RNAs (sgRNAs) that Cas9 needs to target a gene of interest. Cas9-assisted genome editing works in crops too, including maize, but few studies have been brave enough to scale up the analysis by generating and analyzing thousands of transformants. Liu et al. found the courage to step up to the challenge.
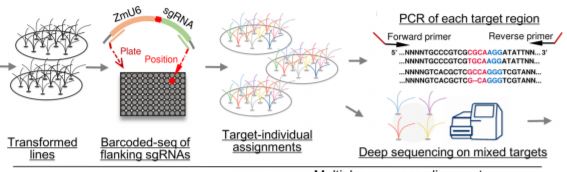
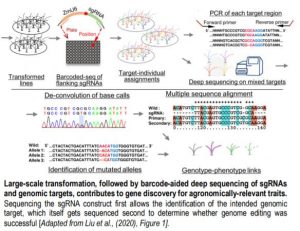
They first integrated multiple sources of information (mutants, GWAS, QTLs) to draw a list of over 1,000 genes with high potential return value to be genome-edited. The authors then cloned the corresponding sgRNAs into transformation constructs by overlap-PCR or ligation of oligonucleotides, and made sure that the sgRNA target site was not polymorphic (a potential issue since they used a maize inbred line distinct from the B73 reference). Rather than maintaining individual clones, they pooled them ahead of Agrobacterium-mediated transformation of maize immature embryos and selected close to 4,400 antibiotic-resistant plants. The identification of the constructs that had inserted into the genome came next by amplifying and sequencing each sgRNA from genomic DNA.
Of course, a positive transformant does not mean that the gene of interest was itself edited, but this can be checked by amplifying the region surrounding the target of the sgRNA and subjecting all barcoded products to deep-sequencing (see Figure). The authors spent time to optimize this step of the analysis, which determines whether a specific gene had been edited or not. And this is where the fun begins. Through typical attrition during large-scale projects, not all 1,000 genes were edited, but 118 that were came back with clear phenotypes. And a 10% return on investment is actually really good. Notable examples shared by the authors include the deconvolution of two distinct classic flowering time QTLs into not one, but two causal loci each within the interval obtained from fine-mapping. Another example provides a word of caution when edited lines have no phenotype: perhaps it might be wise to check the expression of homologous genes, in case the loss of the first gene might not have caused an up-regulation of the others.
Other laboratories may choose a scaled-down version of the pipeline described by the authors to finally nail down that long-sought QTL, or go after all the members of a gene family. Regardless, large-scale screens with libraries of sgRNAs are finally within reach of the plant community.
Patrice A. Salomé
Science Editor
ORCID: 0000-0003-4452-9064
References:
Liu et al., (2020). High-Throughput CRISPR/Cas9 Mutagenesis Streamlines Trait Gene Identification in Maize. Published February 2020. DOI: https://doi.org/10.1105/tpc.19.00934