The Future is Now: Gene Expression Dynamics at Single Cell Resolution
For the past 10 years, a popular method to study global gene expression changes has been RNA sequencing of bulk tissues. However, an inherent limitation of this approach is the confounding of multiple cell types or multiple developmental stages. By contrast, new technologies enable transcriptional profiling of thousands of cells at single cell resolution. Single cell RNA-sequencing (scRNA-seq) of human and animal tissues has produced unprecedented insight into gene expression changes underlying development and response to stimuli. However, until recently, scRNA-seq has never been applied to plants.
Using the 10X Genomics platform, Jean-Baptiste et al. (2019) establish a methodological framework to produce and analyze high-throughput, droplet-based scRNAseq data from Arabidopsis root samples. To test the ability of scRNA-seq to identify known and novel cell types, the authors transcriptionally profiled over 3,000 cells from whole root samples. Similarities in gene expression profiles were used to partition cells into discrete clusters. To determine the cell type represented by each cluster, the authors report the results of three complementary analyses based on published tissue-specific and cell stage-specific gene expression data. Exploration of the clusters revealed hundreds of new genes, including transcription factors, with cell type- and tissue-specific expression profiles. Among these genes are likely candidate regulators of cell identity and differentiation.
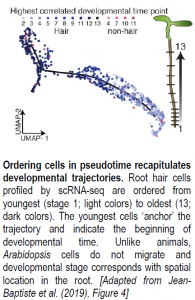 After assigning cell types to each cluster, Jean-Baptiste and colleagues sought to explore the expression dynamics underlying cell maturation. To this end, a particularly exciting application of scRNA-seq is the ability to order cells along a developmental timeline based on overlapping gene expression profiles (see Figure). Cascades of genes differentially expressed between developmental states along the timeline should contain candidate regulators of cell maturation events. Here, the authors used an advanced machine learning approach available in Monocle, a popular R package, to order root hair, endodermis, and cortex cells from young to old in ‘pseudotime.’ Genes differentially expressed between cells at the root of the developmental trajectory (young cells) and those at the end of the trajectory (old cells) for each tissue type corresponded to stage-specific markers previously identified by RNA-seq, suggesting that developmental trajectories can be successfully recapitulated for root cells. Notably, an analysis of gene expression changes over time using an “RNA Velocity” package revealed that transcriptional rates of hair cell-specific genes increased in hair cells as ploidy increased, even as overall RNA levels decreased. These observations are consistent with hair cells becoming more specialized over time as they move toward terminal differentiation. Together, these results highlight the power of scRNA-seq to explore transcriptional dynamics and regulation of developmental events at an unprecedented resolution.
After assigning cell types to each cluster, Jean-Baptiste and colleagues sought to explore the expression dynamics underlying cell maturation. To this end, a particularly exciting application of scRNA-seq is the ability to order cells along a developmental timeline based on overlapping gene expression profiles (see Figure). Cascades of genes differentially expressed between developmental states along the timeline should contain candidate regulators of cell maturation events. Here, the authors used an advanced machine learning approach available in Monocle, a popular R package, to order root hair, endodermis, and cortex cells from young to old in ‘pseudotime.’ Genes differentially expressed between cells at the root of the developmental trajectory (young cells) and those at the end of the trajectory (old cells) for each tissue type corresponded to stage-specific markers previously identified by RNA-seq, suggesting that developmental trajectories can be successfully recapitulated for root cells. Notably, an analysis of gene expression changes over time using an “RNA Velocity” package revealed that transcriptional rates of hair cell-specific genes increased in hair cells as ploidy increased, even as overall RNA levels decreased. These observations are consistent with hair cells becoming more specialized over time as they move toward terminal differentiation. Together, these results highlight the power of scRNA-seq to explore transcriptional dynamics and regulation of developmental events at an unprecedented resolution.
In addition to addressing developmental questions, scRNA-seq is a promising technique for characterizing cellular heterogeneity in response to environmental signals or stress. By exposing roots to high temperatures, this study reports expression changes of both canonical and novel genes among multiple cell and tissue types. Future scRNA-seq applications could therefore include identification of cell types that most strongly respond to different biotic or abiotic stresses. This level of resolution could inform selection of specific cell types for targeted gene manipulation, thereby circumventing pleiotropic effects.
Overall, the agreement of this study’s findings with published Arabidopsis root data supports the validity and reliability of scRNA-seq in an established plant model. Notably, Jean-Baptiste and colleagues demonstrate the exciting power of scRNA-seq to explore total mRNA dynamics across developmental time in specific cell types. Three recent and complementary studies also report successful scRNA-seq experiments with Arabidopsis roots (Shulse et al. 2018) and illuminate systems-level transcriptional perturbations in mutants (Denyer et al., 2019; Ryu et al., 2019). Together, these studies provide a robust framework for analyses in Arabidopsis and support the applicability of scRNA-seq to less well-characterized plants, including crop species.
REFERENCES
Denyer T, Ma X, Klesen S, Scacchi E, Nieselt K and Timmermans MCP. (2019). Spatiotemporal developmental trajectories in the Arabidopsis root revealed using high-throughput single-cell RNA sequencing. Dev. Cell 48: 840-852.e5.
Jean-Baptiste K, McFaline-Figueroa J, Alexandre CM, Dorrity MW, Saunders L, Bubb KL, Trapnell C, Fields S, Queitsch C and Cuperus JT. (2019). Dynamics of gene expression in single root cells of A. thaliana. Published March 2019, DOI: https://doi.org/10.1105/tpc.18.00785.
Ryu KH, Huang L, Kang HM and Schiefelbein J. (2019). Single-cell RNA sequencing resolves molecular relationships among individual plant cells. Plant Physiol. Published February 2019, DOI:10.1104/pp.18.01482
Shulse CN, Cole BJ, Turco GM, Zhu Y, Brady SM, Dickel DE. (2018). High-throughput single-cell transcriptome profiling of plant cell types. bioRxiv. DOI: http://dx.doi.org/10.1101/402966.

