Chasing Scattered Genes: Identifying Specialized Metabolite Pathway Genes through Global Co-expression Analysis
 Plants produce scores of specialized metabolites (SMs) to attract or repel the organisms around them and to cope with life in a variable environment. For thousands of years, we have been exploiting these compounds to feed, heal, and adorn us. Many more SMs remain to be discovered: the chemical constituents of only 15% of the estimated 350,000 plant species on Earth have thus far been explored (Wurtzel and Kutchan, 2016). Since SMs are not required for plant growth or reproduction, the underlying genes and pathways leading to their production have diversified greatly over time and are not well conserved among species, making them difficult to identify through standard homology searches. However, genes within an SM pathway can be identified through their shared regulatory network, since the successful production of an SM requires the underlying genes to be expressed at the right time and place. Searches for co-expressed genes from global gene expression data have shed light on SM pathways in various plants, but technical constraints and limited data have hampered such analyses.
Plants produce scores of specialized metabolites (SMs) to attract or repel the organisms around them and to cope with life in a variable environment. For thousands of years, we have been exploiting these compounds to feed, heal, and adorn us. Many more SMs remain to be discovered: the chemical constituents of only 15% of the estimated 350,000 plant species on Earth have thus far been explored (Wurtzel and Kutchan, 2016). Since SMs are not required for plant growth or reproduction, the underlying genes and pathways leading to their production have diversified greatly over time and are not well conserved among species, making them difficult to identify through standard homology searches. However, genes within an SM pathway can be identified through their shared regulatory network, since the successful production of an SM requires the underlying genes to be expressed at the right time and place. Searches for co-expressed genes from global gene expression data have shed light on SM pathways in various plants, but technical constraints and limited data have hampered such analyses.
In addition to their tight regulation, genes in SM pathways, at least in bacteria and fungi, are often clustered together in the genome, forming biosynthetic gene clusters (BGCs). Powerful tools are used to identify BGCs and to predict their involvement in SM pathways. While most known plant SM pathway genes are dispersed across the genome, several plant BGCs have been identified and many more have been predicted (Schlapfer et al., 2017), and the idea that SM pathway genes in plants tend to be clustered together has been gaining traction. If this is not the case, however, techniques for finding plant SM genes based on chromosomal proximity, an easy-to-detect feature, would fail to uncover most SM pathways, prompting Wisecaver et al. (2017) to investigate this issue using data from eight model plant species.
Based on the assumption that genes in an SM pathway form tightly associated co-expression modules, the authors used pairwise measurements of gene co-expression data from hundreds to thousands of experiments to construct MR (mutual ranks)-based co-expression networks. Genes were then assigned to modules of tightly co-expressed genes using the ClusterONE tool. Focusing on small (<50 gene) modules to reflect the typical size of an SM pathway, the authors looked for modules containing SM pathway genes in the Pfam database, finding the fewest such modules in the green alga, Chlamydomonas reinhardtii and the most in the mustard, Brassica rapa.
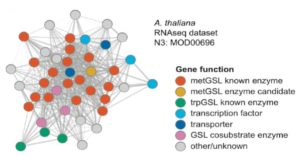
Many (15.3–52.6%) modules contained two or more known SM biosynthetic genes and genes enriched in SM-related functional categories, as well as many experimentally validated SM pathways. For example, this analysis identified almost all genes involved in the methionine-derived aliphatic glucosinolate (metGSL) biosynthesis pathway and associated biochemical processes in Arabidopsis thaliana (see figure). This approach also revealed all six functionally characterized SM pathways known to form BGCs in the eight plant genomes examined. However, an examination of predicted but not experimentally validated BGCs suggested that these clustered genes are not coexpressed and do not form co-expression modules and might therefore not correspond to functional SM pathways after all. Thus, proximity might not be a reliable index for identifying SM pathways, since most are likely scattered, not clustered. Instead, SM pathways manage to produce their highly coveted products through coordinated expression of their genes, a trait that can now be exploited to facilitate their discovery.
IN BRIEF by Jennifer Lockhart jlockhart@aspb.org
REFERENCES
Schlapfer, P., Zhang, P., Wang, C., Kim, T., Banf, M., Chae, L., Dreher, K., Chavali, A.K., Nilo-Poyanco, R., Bernard, T., Kahn, D., and Rhee, S.Y. (2017). Genome-wide prediction of metabolic enzymes, pathways and gene clusters in plants. Plant Physiol: 173: 2041-2059.
Wisecaver, J.H., Borowsky, A.T., Tzin, V., Jander, G., Kliebenstein, D.J., and Rokas, A. (2017). A global co-expression network approach for connecting genes to specialized metabolic pathways in plants. Plant Cell 29: doi:10.1015/tpc17.00009.
Wurtzel, E.T. and Kutchan, T.M. (2016). Plant metabolism, the diverse chemistry set of the future. Science 353: 1232–1236.









Leave a Reply
Want to join the discussion?Feel free to contribute!